|
As evidence mounts for a causal role in the autism epidemic, letter urges NIH committee to prioritize research on heritable (germline) impacts of modern general anesthetic gases. The following letter was submitted to the NIH Interagency Autism Coordinating Committee, a federal advisory committee that coordinates federal efforts and provides advice to the Secretary of Health and Human Services on issues related to autism.
July 1, 2021 To the Members of the IACC: I am writing to urge that your committee recommend federal funding to investigate a question of urgent importance in autism etiology: the genetic and epigenetic impacts of halogenated anesthetic gases on the germ cells (egg, sperm, and lineage of precursors) of the parent generation, and the ensuing impacts on offspring neurodevelopment, in particular the early transcriptional dysregulation of brain-related genes. Last year I co-authored a commentary on this topic, "General anesthesia, germ cells and the missing heritability of autism: an urgent need for research," in the respected peer-reviewed scientific journal Environmental Epigenetics, with Jill Escher, a well-known autism research philanthropist who has been raising questions about this and related matters for many years (Escher and Ford 2020). I am a retired anesthesiologist and mother of a young man, Connor, who has a severe form of idiopathic autism. Based on my experience as a physician with extensive experience in clinical practice and my keen interest in autism causation research, I can say with confidence that this hypothesis, which has gained enthusiastic attention from molecular biologists, germ cell toxicologists, and reproductive biologists, is likely the most important one yet raised in the history of autism. Allow me to explain as best I can, briefly, and in layperson's terms. Every year more than 50 million inpatient surgeries are performed in the United States, including procedures on pregnant women, neonates, young children, older children, adolescents and pre-conception adults. Since the 1960s, my field has adopted waves of new agents to induce the state of anesthesia, including but not limited to intravenous agents like propofol and halogenated anesthetic gases such as halothane, enflurane, isoflurane, desflurane and sevoflurane (which I will call "GA"). These drugs are nearly miraculous in their ability to induce global suppression of the nervous system in the patient so as to enable the myriad procedures of modern surgery (anything from neonatal hernia repair, to plastic surgery, to heart surgery, to orthopedic surgery, to appendectomies, among countless others). That's the good news. The bad news is that these agents are highly toxic. In their ordinary doses they are of course lethal, with patients kept alive only via intubation and careful monitoring. They are neurotoxic-- in fact, the FDA has issued a warning regarding neurotoxicity in patients under the age of three. They are genotoxic — seen to induce DNA damage at the site of contact and peripheral blood. They are reproductive toxicants -- which can damage sperm and egg. More importantly for our purposes, a rash of new studies in mammal models show they can induce epigenetic changes in germ cells that can result in abnormal neurodevelopment in the offspring. These papers are cited in our Environmental Epigenetics paper, and I will not repeat them now, except to add that a remarkable newly published study in Biology of Reproduction has shown how fetal exposure to sevoflurane can induce abnormal binding of transcription factors in sperm DNA, resulting in dysregulated transcription of autism-related genes in offspring -- and abnormal brain and autism-related behavioral phenotypes (Wang et al. 2021). In other words, it appears, based on animal studies, that germ cell exposure to GA agents can exert an adverse intergenerational impact. In hindsight, this hypothesis strikes most people as rather obvious. And indeed researchers first noticed these heritable effects via mouse experiments in the early 1980s. But regrettably, no heritable-effect studies were performed again until a few years ago (Escher and Ford 2020), and the FDA does not test for germline impacts of GA. As a practitioner neither I nor my colleagues gave any consideration to our patients' germ cells. This facet of GA toxicology has languished in a blind spot for nearly 4 decades. While I believe that surgeries under GA are likely benign to germline in most cases, I am deeply concerned about certain exposures, for example, exposures to GA in infancy and childhood, as well as repeated or prolonged exposures, including in adulthood. And while GA likely poses little absolute risk to a patient's germ cells, the population level risk may be substantial. If just 1% of U.S. patient exposures result in an adverse heritable effect, that could result in 500,000 cases of some level of abnormal neurodevelopment per year. Through the years I have watched many hypotheses of autism causation come and go. The current trend is to presume autism's strong heritability "genetic" -- an idea that seems sensible on its face but is lacking in actual molecular evidence except with regards to rare de novo germline mutations which can only explain a fraction of the cases. And while I have no way of knowing what subsection of autism cases may be explained by heritable impacts of parental exposures to GA, I must point out the unprecedented power of this hypothesis to explain a great number of unsolved mysteries of autism, including the following: --The tremendous increase in prevalence in recent decades, particularly in industrialized countries --Autism's strong heritability (via broad impacts across parental germ cells) --Strong sibling recurrence --The genesis of autism in early abnormalities of transcription of brain-related genes, impacting neurogenesis, neural migration, and synaptic function --The heterogeneity of autism spectrum disorders --The skewed male-to-female ratio (males are markedly more affected in the mammalian experiments) --Regional and demographic variations in prevalence --The "broader autism phenotype" seen in parents and siblings The IACC was created to push federal agencies to finally answer questions about autism, and I can think of no other question in autism research that deserves more attention than this. And this attention is 40 years overdue. If the committee wishes to learn more about this important emerging hypothesis, please do not hesitate to contact me or my co-author. Thank you for your attention. Respectfully, La Donna Ford, MD Foster City, California [email protected] References: Escher J, Ford LD. General anesthesia, germ cells and the missing heritability of autism: an urgent need for research, Environmental Epigenetics 2020;6(1), dvaa007, https://doi.org/10.1093/eep/dvaa007 Wang HL, Forestier S, Corces VG. Exposure to sevoflurane results in changes of transcription factor occupancy in sperm and inheritance of autism. Biology of Reproduction 2021;ioab097, https://doi.org/10.1093/biolre/ioab097
0 Comments
[I sent this letter (PDF here) to the NIH following on a correspondence of November 2018, which you can find here. It again exhorts the NIH to specifically fund research on this extraordinarily crucial issue. You can read the response by Dr. Bianchi here. —Jill Escher] Francis Collins, MD, PhD Director National Institutes of Health Building 1, Room 118A 1 Center Drive Bethesda, MD 20814 Joshua Gordon, MD, PhD Director National Institute of Mental Health Building 31, Room 4A52J 31 Center Drive Bethesda, MD 20814 Linda Birnbaum, PhD Director National Institute of Environmental Health Sciences Building 101, Room B242 111 T W Alexander Drive Research Triangle Park, NC 27709 Diana Bianchi, MD Director National Institute of Child Health and Human Development Building 31, Room 2A03 31 Center Drive Bethesda, MD 20814 Via Email and U.S. Mail June 21, 2019 Re: NIH research program on heritable (germline) impacts of general anesthesia, a response to Dr. Bianchi’s November 27, 2018 letter Dear Dr. Collins and Institute Directors: On November 4, 2018 I took the liberty of writing to you suggesting a research program on the adverse heritable impacts of general anesthesia (GA) (Escher 2018b). On November 27, 2018 Dr. Bianchi kindly responded, expressing skepticism about the hypothesis and rejecting the idea that the question warranted any particular action by the NIH (Bianchi 2018). She did allow, however, that the NIH would consider investigator-initiated grant applications proposing studies on this topic (but, alas, I have it on good information that when such an application was recently made it was met with rejection).(1) I am now asking you to reconsider the NIH’s response to my earnest suggestion. This is not me saying, “Please spend a zillion dollars studying mating habits of fruit bats,” but instead, “Rates of autism —a highly heritable but not classically genetic disorder — have hit a catastrophic 1 in 59 U.S. children, please, at the very least, consider a modest sum to investigate the heritable impacts of the most toxic exposure our germ cells commonly encounter, one with proven adverse heritable neurodevelopmental impacts.” I strenuously disagree with Dr. Bianchi’s assertion that “At present, there are limited scientific data to support your hypothesis,” as even a casual glance at the research literature reflects not just the unmistakable plausibility for this idea, but also the tremendous urgency of the issue. GA is genotoxic and a powerful modifier of chromatin. It is a germ cell toxicant that is known to dysregulate the expression of brain development genes. And moreover, when volatile inhalation anesthetic gases are actually tested for heritable impacts via germ cell exposure, neurobehavioral abnormality is the result seen in animal models, and with a male-affected bias (as Dr. Bianchi noted in her letter). But molecular plausibility is just the beginning, as the hypothesis also features strong congruence with abundant findings in autism research, including temporal and prevalence trends, heritability patterns, neurobiology and epidemiology. I will not repeat any of the red-flag family stories as I did to an extent in my November 4 letter. But I can assure you that just one single individual seemingly impaired by germ cell anesthesia toxicity can easily cost the family and public $10 million over a lifetime. In light of the astronomical costs that may be resulting in part from GA-damaged germline, is it too much to ask the NIH to sponsor, say, $1 million in pilot research? What are these germline toxicants doing to our germ cells now, and how much heritable damage have they done since their introduction in the late 1950s?(2,3,4) There is only one way to find out. And that is a robust research program.(5) With that goal in mind I will explain what the studies are already telling us, and why they compel us to do much more. 1. GA gases can penetrate germ cells and enter the nucleus, where they can damage DNA and chromatin The GA gases are potent, small lipophilic molecules that, on an organismal level, are lethal in their conventional doses: patients, whose brain and muscle function shut down, are kept alive only through a breathing apparatus and careful monitoring. The gases are basically powerfully poisonous organic solvents which diffuse through the body, particularly vessel-rich tissues such as the gonads, and every aspect of the cell, dramatically impacting receptors, signaling, chromatin, and even DNA. The gas gets free passage into the nucleus and all its structures including DNA, its support proteins, and epigenetic elements. When a body is anesthetized, so in effect are its germ cells and their heritable components. Once the GA gases enter the germ cells, then what?(6) To a large extent this will depend not only on the pharmacokinetics of the particular substance but also on combination, dose, and timing. Timing is a critical variable for germ cells owing to enhanced vulnerabilities during early epigenetic reprogramming, chromatin remodeling, rapid mitotic proliferation, early stages of meiosis, genomic imprinting, and spermiogenesis in males and meiosis II in females.(7) It is beyond dispute that a transient exposure during a critical period of germline remodeling can cause changes that become fixed in the genetic apparatus, resulting in the dysregulation of gene expression and proper and timely neuronal development in progeny, a phenomenon already observed not only with respect to GA (Ju et al. 2018), but also overtly hormone-disrupting drugs such as diethylstilbestrol, synthetic corticosteroids, hormone-disrupting environmental chemicals, valproic acid, tobacco, nicotine and ethanol (reviewed in Escher and Robotti 2019). This phenomenon occurs because exogenous toxicants can act directly or indirectly to effectuate alterations in DNA methylation, histone modification and/or ncRNA expression in the germ cells (Marczylo et al. 2016; Gold et al. 2018; Western 2018), or, with respect to some exposures like tobacco, outright mutagenesis (DeMarini 2012). Today, up to 75,000 pregnant women undergo inpatient surgical procedures each year, exposing the fetal germ cells.(8) Additionally, 6 million children, 1.5 million of whom are infants, also undergo surgery (Gluncic et al. 2019), exposing their nascent germ cells. Of additional concern is that GA gases are used in higher concentrations in infants under the age of one year, and in particularly high concentrations in maternal-fetal medicine for antenatal corrective surgeries (to relax the uterus, intensive concentrations of sevoflurane are used for many hours, exposing a very undeveloped fetus to unprecedented quantities of GA). Beyond the question of timing is that of dose and recurrence. GA agents are used in varying combinations and concentrations, and for varying durations, depending on the judgment of the practitioner, the availability of the chemicals, and the demands of the surgical procedures. Often a condition requires several successive surgeries, resulting in a cumulative exposure that could exacerbate damage or derail the chromatin and DNA repair process in the GA-exposed nucleus. In sum, GA gas is potent and penetrant, even to the germ cell nucleus, but it is timing, dose, and repetition that may “make the poison” to the exposed germ cells.(9) 2. GA is a powerful modulator of epigenome and chromatin GA gases have been shown to be powerful modulators of chromatin remodeling and epigenetic function that induce a wide variety of morpho-functional effects when administered during critical periods of brain development (Vutskits et al. 2018; Bang 2015). The injury often occurs via changes in key transcription factors leading to dysreguation of gene expression necessary for normal neurodevelopment (Vutskits et al. 2018; Csoka and Szyf 2009). From a neurological point of view, damage includes neuronal apoptosis, impairments in synaptogenesis and defects in neuronal migration, leading to ectopic neurons (Gluncic et al. 2019). Adverse outcomes observed have included learning impairments, brain abnormalities, behavioral abnormality (Id). Research finding epigenetic and transcriptional impacts in brain cells date back to several studies published 2006. In cultured neurons, isoflurane altered several genes involved with neurotransmitter transport, signaling and cellular structure (Pan et al. 2006). Isoflurane was also seen to affect widespread changes in genetic control in the rat amygdala, and alter gene expression related to DNA transcription, protein synthesis, metabolism, signaling cascades, cytoskeletal structural proteins, and neural-specific proteins, among others (Rampil et al. 2006). Isoflurane with nitrous oxide caused persistent changes in hippocampal gene expression in rats. The majority of differentially expressed genes are implicated in cell stress and replication, signal transduction, transcription, protein biosynthesis, cell structure, and metabolism (Culley et al. 2006). Later studies confirmed and expanded on these findings. In rats, isoflurane altered hippocampal protein expression, affecting processes including synaptic plasticity, stress response, detoxification, and cytoskeleton (Kalenka et al. 2010). In mice, even a brief exposure to isoflurane persistently upregulated expression in several genes in the hippocampus (Pekny et al. 2014). Neonatal sevoflurane anesthesia (in combination, mirroring clinical reality) in rats was found to produce long-lasting alterations in histone acetylation, resulting in impairments of hippocampal synaptic plasticity: reduced density of dendritic spines, reduced levels of the brain-derived neurotrophic factor, c-fos protein, microtubule-associated protein 2, synapsin1, postsynaptic density protein 95, pCREB/CREB, CREB binding protein, and acetylated histones H3 and H4, and increased levels of histone deacetylases 3 and 8 (Jia et al. 2016). A cascade of events was seen to be initiated by sevoflurane-induced epigenetic modulations. By promoting CBP degradation, the gas induced significant down-regulation of full-length CBP protein, which resulted in a decrease in its HAT activity. This, in turn, caused H3 hypoacetylation, an epigenetic change that leads to more condensed chromatin structure less conducive to transcription of the target genes BDNF and c-Fos, which are critical for cognitive development. An impairment in proper dendritic arborization leads to impaired neuronal connectivity resulting in faulty formation of neuronal circuits and compromised synaptic neurotransmission (Jia et al. 2016). The GA isoflurane, with associated sedatives, can modulate histone acetylation and as such may have deleterious effects on transcription of genes crucial for proper synapse formation and cognitive development. During synaptogenesis, epigenetic changes have been shown to involve key transcription factors (e.g. cAMP response element-binding [CREB] protein, CREB-binding protein) leading to downregulation of target genes (e.g. brain-derived neurotrophic factor, c-Fos) via histone modification (Dalla Massara et al. 2016). Most recently, neonatal sevoflurane-induced alteration of brain gene expression, modulation of KCC2 gene expression via modification of DNA methylation, was noted in rats (Ju et al. 2018), along with adverse impacts on progeny gene expression and neural function (discussed later in this letter). A study looking at isoflurane effects on migration of cerebral cortical neurons revealed that significant number of neurons failed to acquire their correct cortical position and remained dispersed within inappropriate cortical layers and/or adjacent white matter, linked to diminished expression of proteins critical for neuronal migration. Behavioral abnormalities in exposed offspring were also noted (Gluncic et al. 2019). Remarkably, the neuronal defects induced by developmental GA exposure, such a abnormal migration of cortical neurons and impaired synaptogenesis, mirror the impairments seen in post-mortem autism brains (Hutsler and Casanova 2015; Reiner et al. 2015), raising questions about dysregulation of the associated genes via the autistic subject’s germline. It stands to reason that the neurodevelopment genes targeted by GA gases in brain cells would also be targeted in germ cells. But unlike differentiating neurons, a lesion at the blueprint germ cell level could potentially lead to more acute phenotypic consequences due to systemic interference with the precise tempo-spatial processes of brain development. 3. GA gases are also genotoxic, causing DNA damage Data also indicate that GA is associated with genotoxic risks (reviewed in Yilmaz et al. 2016; Schifilliti et al. 2011; discussed in Çakmak et al. 2018), although those risks seem to be agent and dose dependent. Evidence shows that inhalation anesthetics cause DNA damage in a variety of cells: at site of contact (epithelial cells in the nose) and systemically (blood cells). At contact sites, short-term administration of sevoflurane was seen to induce micronucleus formation in nasal epithelial cells of patients (Kesimci et al. 2017). In blood, a comet assay detected DNA damage caused by halothane and isoflurane in human peripheral blood lymphocytes (PBLs) (Jaloszynski et al. 1999). In human patients, desflurane increased sister chromatid exchange in lymphocytes (Akin et al. 2005). The comet assay of halothane and desflurane were shown to be genotoxic in lymphocytes, increasing DNA migration in a dose-dependent manner (Karpinski et al. 2005). Dose-related genotoxicity of desflurane in lymphocytes was again observed as shown by comet assay (Aydinli et al. 2011). Consistent with a dose-dependent effect, in minimally invasive surgery, isoflurane and sevoflurane were not seen to induce DNA strand breaks or alkali-labile sites in PBLs (Braz et al. 2011). Halothane, isoflurane, sevoflurane and desflurane were investigated in human PBLs (and sperm cells also, discussed later) in vitro by alkaline comet assay. All drugs were capable of inducing DNA damage on PBLs in a dose-dependent manner (Kaymak et al. 2012). In minor surgeries, desflurane caused statistically significant increases in DNA strand breaks/alkali-labile sites in lymphocytes the day after minimally invasive surgery in healthy patients (Nogueira et al. 2016). 4. GA gases are also cytotoxic to germ cells In case there is any question that GA gases can reach and impair germ cells, it is widely observed that GA agents have generally deleterious impacts on morphology and integrity of mammalian and human germ cells. Sperm/males Morphologic changes in mouse sperm caused by various forms of anesthesia was first discussed in 1981 when significant increases in the percentages of abnormal spermatozoa were found for chloroform, trichloroethylene, and enflurane (Land et al. 1981). “These data suggest that direct examination of reproductive cells following exposure to general anesthetics in vivo may be useful in the investigation of the genetic toxicities of these compounds” (Id). Halothane produces an inhibition of rat masculine sexual behavior and reduced sperm motility (Oropeza-Hernández et al. 2002). Although not a halogenated volatile anesthetic, the inhalatory anesthetic ethyl ether during the neonatal period of brain sexual differentiation impaired later fertility and sexual behavior of male rats: a decrease in the number of spermatids and spermatozoa, an increase in the transit time of cauda epididymal spermatozoa and a decrease in daily sperm production. An alteration of sexual behavior was also observed. This may be because perinatal exposure to ethyl ether acting as a hormone disruptor during the critical period of male brain sexual differentiation (Arena et al. 2002). In rabbits, exposure to sevoflurane and isoflurane had negative effects on spermatogenesis and sperm morphology, concentration and motility (Ceyhan et al. 2005). An in vitro study of human sperm showed that isoflurane has a reversible increasing effect at the clinical concentration and a significant decreasing effect at the high concentration on the motility and vitality of sperm, while sevoflurane does not affect sperm motility and vitality at either concentration (Wang et al. 2008). Isofurane impaired rat seminiferous tubules and spermatogenesis, damage related to the alterations of sex hormones (Xu et al. 2012). In rats, sevoflurane damaged testicular and sperm morphology, and reproductive hormones were affected by chronic exposure (Kaya et al. 2013).(10) Oocytes/females As with most aspects of germ cell toxicology, information about the fate of oocytes is rare compared to the male gametes, but two studies suggest the germ cell toxicant impacts of the gases. In a study comparing rat euthanasia methods, euthanasia by isoflurane resulted in significantly fewer intact oocytes in females compared to those killed by cervical dislocation (Roustan et al. 2012). Sevoflurane in female rats results in significant histological ovarian injury and significant alterations in hormone levels (Dogru et al. 2017).(11) 5. GA can exert adverse heritable impacts Most importantly, research has directly demonstrated the adverse heritable impacts of GA. After Land et al. first noted the germ cell toxicity caused by enflurane in 1981, the lab of Herman Turndorf, MD at NYU questioned whether adulterations in the germline could cause heritable impairment. Turndorf’s lab demonstrated that both halothane and enflurane exposed germ cells resulted in learning-impaired progeny (Chalon et al. 1981; Tang et al. 1985). They observed that grandpups of female mice exposed to halothane as fetuses exhibited impaired learning due to what they perceived as a "genetic aberration” in the exposed mothers’ fetal eggs (Chalon et al. 1981). Then again they found impaired learning function in the generation borne of enflurane-exposed mouse sires, prompting them to state that it “seems likely that spermatogenetic changes, caused by enflurane, are associated with genetic alterations” that affected the pups’ brain development (Tang et al. 1985). These crucial observations seemed to go into hibernation for more than three decades but were discussed again in Jia et al 2016 and then actually demonstrated again in Ju et al. 2018, which found that male, but not female, progeny showed signs of neurodevelopmental impairment induced by germline exposure to sevoflurane (Ju et al 2018).(12) In sum, the mammal studies so far published on this question all point in one direction — that germline exposure to GA can cause learning and behavioral impairment in progeny. Relevance of the germline toxicity of GA to the autism increase in particular was first raised by Escher in 2018 and in Ju et al. 2018, a hypothesis repeated by Escher and Robotti in 2019, emphasizing the need for a rear-view-mirror approach to ascertaining heritable pathologies unwittingly induced by the historical growth in use of novel GA agents and other potent drugs developed in the post-war decades. 6. Consistency with findings from autism research While studies indicate that GA can enter the nucleus, derange chromatin, epigenome and DNA, cause gametic abnormality, and induce adverse neurodevelopmental outcomes in progeny, what is equally striking that this historic-biologic phenomenon could explain many of the baffling patterns seen in the autism research literature. Here are some examples: Temporal associations. The start of the autism increase, observed to have begun with births in the early 1980s (Nevison et al. 2018), comes roughly a generation after early germ cell exposures to synthetic volatile inhalation gases (starting with halothane in the late 1950s, which remained the most prevalent GA for decades). Missing heritability of autism. The epigenetic / chromatin / genomic effects of GA could help explain the contrast between the strong heritability of autism and the surprisingly shallow findings from traditional DNA-sequence-focused genetics. The 4:1 male:female sex ratio. The hypothesis is consistent with the sex-specific intergenerational responses to GA exposure as detected in Ju et al. 2018. Additionally, several studies in chemical disruption of germ cells have found male offspring more likely suffer adverse effects (e.g., Krishnan et al. 2018). Autism heterogeneity. Toxicant exposures to male or female germ cells over different times, in different doses, in different combinations, against a backdrop of varying genomic susceptibilities and different sexes, would likely exert widely variable effects. This roulette-wheel mix could help explain the heterogeneity of the autisms. The “broader autism phenotype.” The BAP has been observed among autism family members. In many cases, personalities and cognitive traits of parents themselves could have been influenced by their direct in utero or early life exposures to neurotoxic GA, and in addition, siblings who do not meet diagnostic criteria for autism could have sprung from germ cells that were more lightly damaged. Parental age effects. It has often been noted that paternal and also maternal age is associated with offspring autism risk. One reason for this phenomenon, apart from rare random mutation of nucleotide sequence in the germline, could be the higher rates of toxicant exposure experienced by the parents over the pre-conception lives. Cumulative exposure may confer greater heritable risks (Gao et al. 2019). Regional, socioeconomic, and ethnic disparities. Higher rates of autism in some countries, regions, ethnicities and socioeconomic strata may coincide with higher rates of GA exposures of the parents. Surgery is more prevalent in some countries and demographics than others. Arising in early brain development. It has been frequently observed that autism arises from brain mis-wiring during early development in the womb. Increasingly it looks like chromatin and epigenomic factors may contribute, suggesting that “epigenetic dysfunction is a fundamental contributor to brain development and disease pathogenesis of neurodevelopmental disorders, including ASD” (Tremblay and Jiang, 2019). The dysregulation of brain development genes induced by GA germline exposure could help explain these phenomena. A Priority for NIH Research In sum, upon examination of the literature it is not difficult to connect the dots between germline exposure to agents of general anesthesia and heightened risk for progeny neurodevelopmental impairment. As we witness a baffling tsunami of young Americans with serious functional and behavior impairments — disorders shown to be highly heritable but not strongly genetic in any classic sense — the American public deserves a research program that considers that some of the causes of this catastrophe may lie in the disturbed molecular program of parental germ cells. Several of the NIH Institutes could make this research a priority, both through intramural and extramural programs. For example: NICHD: The NICHD supports both basic and applied research into germ cell health. Some of these funds could be directed to research on germline and heritable toxicity of GA, with emphasis on halothane (most prevalent in the autism parent generation) and sevoflurane (most prevalent today), and with reference to very early life exposures (prenatal and neonatal) and/or long-dose and successive surgeries. NIEHS: The NIEHS also supports both basic and applied research into adverse heritable impacts of exogenous toxicants. Some of these funds could be directed to research on germline and heritable toxicity of GA. For example, rodent models can provide a rough idea of impacts of GA on the next generation’s gene expression, brain function, and behavior. As above, research should begin with the most vulnerable developmental periods (eg, fetal germ cells), the stronger concentrations and longer durations.(13) NIMH: The NIMH is charged with sponsoring relevant research to uncover the causes of autism and other neurodevelopmental disorders. As such, it could direct a research program on a wide variety of retrospective studies across a multitude of human cohorts examining neurodevelopmental outomes in progeny of parents with very early and/or very intensive surgical exposure histories. Of course the FDA should also play a role. However, based on many years of communication with staff at both the regulatory and research (NCTR) sides of the FDA, it is clear that the FDA presently has no program or interest in the adverse heritable consequence of this or any other pharmaceutical drug, with perhaps the exception of chemotherapy drugs, leaving a most vulnerable phase of the human lifecycle virtually orphaned, without any clear “home” among the many institutes mandated to advance research and safeguard public health. For the reasons set forth above, I believe the time has come for the NIH to consider what the most common, intensive germline toxicant exposure has meant and continues to mean for America’s children. Thank you for your consideration of this suggestion. Should you have any questions please contact me at [email protected]. Very truly yours, Jill Escher cc: EPA: David DeMarini FDA: Robert Heflich, William Slikker, William Mattes CDC (NCBDDD): Colleen Boyle NIEHS: Richard Woychik, Cindy Lawler (EEARN) Louis Reichardt, Simon Foundation Thomas Frazier, Autism Speaks Footnotes 1. Imagine that in 1971 proposals to investigate diethylstilbestrol carcinogenicity in humans were rejected owing to lack of human studies demonstrating carcinogenicity. This is the absurd Catch-22 we face today with respect to germline GA exposure studies, even though progeny neurodevelopmental impairment was witnessed in animal models more than 30 years ago. 2. Because most of the GA toxicity research involves the synthetic volatile inhalation gases halothane (introduced to clinical practice in the late 1950s), enflurane (late 1960s), isoflurane (1981), desflurane (1992), and sevoflurane (1992), I will limit my discussion to these. This is not to say that other forms of anesthesia are unimportant, but the volatile inhalation gases are better understood and are alone sufficient to raise concern. 3. I fully appreciate that GA, and in particular the advent of the synthetic volatile inhalation gases, represents one of the greatest medical achievements in history. These substances, though technically destructive and poisonous, have made possible the practice of modern surgery that has saved countless millions of lives. As someone who benefitted from GA for delicate spinal surgery several years ago, my personal gratitude to modern anesthesiology knows no bounds. But our enthusiasm for the technological triumph of GA should not divert us from ascertaining molecular risks which could promote outsize, if unanticipated, developmental havoc. 4. In no way do I suggest that other hypotheses are not worth exploring. For example, adverse neurodevelopmental impacts of early germ cell exposure to smoking (Golding et al. 2017) and drugs such as synthetic steroid hormones (Kioumourtzoglou et al. 2018) stand out as particularly important. Other emerging hypotheses include immune activation and adverse perinatal events, for example. At best, the germline toxicity of some GA exposures would explain a portion of the autism increase. 5. While our philanthropy can help support a few studies (small in vivo, in vitro and epidemiological pilots are now underway), these projects at best will only be able to produce small amounts of pilot data on extremely narrow subparts of this sprawling hypothesis. 6. Another consideration is GA impact on the gonadal somatic support cells, Sertoli and Leydig cells in males, and granulosa and thecal cells in females. If those cells are damaged by gas, the germ cells they support would suffer indirectly. 7. I am addressing what most describe as “intergenerational” effects—those resulting from a direct hit to the germ cell during gametogenesis— and not “transgenerational” effects, which by most definitions are limited to phenotypes that arise in subsequent generations absent any direct germ cell exposure (Miska and Ferguson-Smith 2016; Jarred et al. 2018), even though those effects may pose additional concerns for personal and public health. 8. Prenatal, perinatal, neonatal and early childhood surgeries involving GA in the past or in current practice include maternal appendectomy, maternal injury, cerclage, cesarean section, correction of congenital malformations such as hernias, heart defects, fistulas, clefts or clubfoot, reconstruction after injuries, childhood appendectomies, tumor removal, and many others. As glorious and life-saving these procedures may be (and they are), one cannot deny that evolution did not prepare our delicate early germ cells for biologically unprecedented toxic insults like GA. 9. I have no reason to believe that GA gases are so germline-toxic that they always or even often disturb gametic contents. Based on the literature as a whole, adverse impacts are likely limited to vulnerable developmental periods and/or very heavy and repeated exposures. However, given the extensive and increasing use of synthetic volatile inhalation GA throughout the population over the past six decades, even a five percent increase in heritable risk for abnormal neurodevelopment could silently result in a calamitous, if unforeseen, population-wide impact. 10. The fact that GA acts as an endocrine disrupting chemical, administered in intensive doses, also presents a secondary level of toxic insult to developing germ cells (Marczyclo et al. 2016; De Felici and La Sala 2016; Krishnan et al. 2018). 11. It is worth noting that the gases can be so reproductively toxic to human females that even incidental exposure to waste anesthetic gases can raise the risk of miscarriage in operating room personnel (Yilmaz et al. 2016). 12. With the sole exception of the Ju paper, why did this urgent question fall into the scientific abyss for so many decades? After all, two papers had suggested the deeply troubling prospect of mental impairment in progeny via mysterious “genetic aberrations” or “genetic alterations” of female or male germ cells. Surely, if GA agents could damage our sperm and eggs’ genetic material in a way that caused learning deficits in the next generation, that should have been a top priority for public health research. However, in appears the observations reported by Turndorf’s lab fell victim to the weight of conventional dogma about inheritance. It was broadly accepted at that time that heritability of traits depended on genes from our parents, except in those rare cases where genes suffered a random mutation. The dogma left no room for other ideas about molecular sources of inheritance. Regrettably, even today, pathogenesis research seems paralyzed by old dogmas of the 20th century, with heritability typically equated to “genetic” (Escher and Robotti 2019). 13. In 2018 The Escher Fund nominated the GA gases as a subject for intramural research at the NIEHS National Toxicology Project. The NIEHS response to the nomination is unknown at this time. References
Akin A, Ugur F, Ozkul Y, Esmaoglu A, Gunes I, Ergul H. 2005. Desflurane anaesthesia increases sister chromatid exchanges in human lymphocytes. Acta Anaesthesiol Scand 49:1559–1661. Arena AC, Pereira OC. 2002. Neonatal inhalatory anesthetic exposure: reproductive changes in male rats. Comp Biochem Physiol C: Toxicol Pharmacol 133:633–40. Aydinli B, €Ozg€ok A, Demir ZA, Yagar S, Erg€un MA, Karaer D, Ilhan MN. 2011. Evaluation of dose-related genotoxicity of desflurane by SCE human lymphocytes. Turk J Med Sci 41:1037–1041. Bang SR. 2015. Neonatal anesthesia: how we manage our most vulnerable patients. Korean J Anesthesiol 68(5): 434–441. Bianchi D. Letter to Escher dated November 27, 2018. Archived at: http://www.germlineexposures.org/uploads/6/4/0/9/6409433/bianchiresponse.pdf Braz MG, Braz LG, Barbosa BS, Giacobino J, Orosz JE, Salvadori DM, Braz JR. 2011. DNA damage in patients who underwent minimally invasive surgery under inhalation or intravenous anesthesia. Mutat Res 726:251–254. Çakmak G, Eraydın D, Berkkan A. 2019. Genetic damage of operating and recovery room personnel occupationally exposed to waste anaesthetic gases. Hum & Exper Toxicol 38:1. Chalon J, Tang CK, Ramanathan S, Eisner M, Katz R, Turndorf H. 1981. Exposure to halothane and enflurane affects learning function of murine progeny. Anesth Analg 60:794–7. Ceyhan A, Cincik M, Bedir S, et al. 2005. Effects of exposure to new inhalational anesthetics on spermatogenesis and sperm morphology in rabbits. Arch Androl 51:305–15 Csoka AB, Szyf M. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. 2009. Med Hypoth 73;770–780. Culley DJ, Rustam Y, Xiebe Z, Galic RR, Tanzid RE, Crosby G. 2006. Altered hippocampal gene expression 2 days after general anesthesia in rats. Euro J Pharmacol 549:1-3;71-78. Dalla Massara L, Osuru HP, Oklopcic A, Milanovic D, Joksimovic, SM, Caputo V, DiGruccio MR, Ori C, Wang G, Todorovic SM, Jevtovic-Todorovic V. 2016. General anesthesia causes epigenetic histone modulation of c-fos and brain-derived neurotrophic factor, target genes important for neuronal development in the immature rat hippocampus. Anesthesiology 124:1311-1327. De Felici M, La Sala G. 2016. Epigenetic reprogramming in the mammalian germ line: Possible effects by endocrine disruptors on primordial germ cells. Open Biotech 10:36–41. DeMarini DM. 2012. Declaring the Existence of Human Germ-CellMutagens. Environ Mol Mutagen 53:166-172. Dogru S, Dogru HY, Butun I, Arici A, Benli I, Karaman T, Tapar H, Sahin A, Karaman S, Arici Ozsoy AZ. 2017. Effects of sevoflurane on female reproductive functions in Wistar rats. J Pak Med Assoc 877-883. Escher J. 2018a. Bugs in the program: can pregnancy drugs and smoking disturb molecular reprogramming of the fetal germline, increasing heritable risk for autism and neurodevelopmental disorders? Environ Epigen 4:2;dvy001. Escher, J. 2018b. Letter to NIH Directors re Heritable Effects of General Anesthesia. Dated November 4, 2018. Archived at: http://www.germlineexposures.org/blog/request-for-nih-research-program-on-the-heritable-effects-of-general-anesthesia. Escher J, Robotti S. 2019. Pregnancy drugs, fetal germline epigenome, and risks for next-generation pathology: a call to action. Environ Mol Mutagen, in press. Gao A, Moorjani P, Sasani TA, Pedersen BS, Quinlan AR, Jorde LB, Amster G, Przeworski M. 2019. Overlooked roles of DNA damage and maternal age in generating human germline mutations. PNAS 116 (19) 9491-9500. Gold HB, Jung YH, Corces VG. 2018. Not just heads and tails: The complexity of the sperm epigenome. J Biol Chem 293(36):13815–13820. Golding J, Ellis G, Gregory S, Birmingham K, Iles-Caven Y, Rai D, Pembrey M.l. 2017. Grand-maternal smoking in pregnancy and grandchild’s autistic traits and diagnosed autism. Sci Rep 7:46179. Gluncic V, Moric M, Chu Y, Hanko V, Li J, Luki IK, Luki A, Edassery SL, Kroin JS, Persons AL, Perry P, Kelly L, Shiveley TJ, Nice K, Napier CT, Kordower JH, Tuman KJ. 2019. In utero Exposure to Anesthetics Alters Neuronal Migration Pattern in Developing Cerebral Cortex and Causes Postnatal Behavioral Deficits in Rats. Cereb Cortex, in press. Guo H, Hu B, Yan L, Yong J, Wu Y, Gao Y, Guo F, Hou Y, Fan X, Dong J, et al. 2017. DNA methylation and chromatin accessibility profiling of mouse and human fetal germ cells. Cell Res 27:165–183. Hutsler JJ, Casanova MF. 2015. Cortical construction in autism spectrum disorder: columns, connectivity and the subplate. Neuropath and Applied Neurobiol 42:2;115-134. Jaloszynski P, Kujawski M, Wasowicz M, Szulc R, Szyfter K. 1999. Genotoxicity of inhalation anesthetics halothane and isoflurane in human lymphocytes studied in vitro using the comet assay. Mutat Res 439:199–206. Jarred EG, Bildsoe H, Western PS. 2018. Out of sight, out of mind? Germ cells and the potential impacts of epigenomic drugs. F1000Res 15935.1. Jia M, Liu WX, Yang JJ, Xu N, Xie ZM, Ju LS, Ji MH, Martynyuk AE, Yang JJ. 2016. Role of histone acetylation in long-term neurobehavioral effects of neonatal exposure to sevoflurane in rats. Neurobiol Dis 91: 209-20 Ju LS, Yang JJ, Morey TE, Gravenstein N, Seubert CN, Resnick JL, Zhang JQ, Martynyuk AE. 2018. Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats. Brit J Anesth 121:2;406-416. Kalenka A, Gross B Maurer MH, Thierse HJ, Feldmann RE. 2010. Isoflurane Anesthesia Elicits Protein Pattern Changes in Rat Hippocampus. J Neurosurg Anesthesiol 22:2;144-154. Karpinski TM, Kostrzewska-Poczekaj M, Stachecki I, Mikstacki A, Szyfter K. 2005. Genotoxicity of volatile anaesthetic desflurane in human lymphocytes in vitro, established by comet assay. J Appl Genet 46:319–324. Kaya A, Sogut E, Cayli S, Suren M, Arici S, Karaman S. 2013. Evaluation of effects of repeated sevoflurane exposure on rat testicular tissue and reproductive hormones. Inhalation Toxicology Int Forum for Respiratory Research 25:4. Kaymak C, Kadioglu E, Coskun E, Basar H, Basar M. 2012. Determination of DNA damage after exposure to inhalation anesthetics in human peripheral lymphocytes and sperm cells in vitro by comet assay. Hum Exp Toxicol 31:1207–1213. Kesimci E, Erdem C, Uğur G, Müderris T, İzdeş S, Karahalil B. 2017. Can Sevoflurane Induce Micronuclei Formation in Nasal Epithelial Cells of Adult Patients? Turk J Anaesthesiol Reanim 45(5): 264–269. Kioumourtzoglou M, Coull BA, O’Reilly ÉJ, Ascherio A, Weisskopf MG. Association of Exposure to Diethylstilbestrol During Pregnancy With Multigenerational Neurodevelopmental Deficits. 2018 JAMA Pediatr 172:7;670-677. Krishnan K, Nitish Mittal N, Thompson LM, Rodriguez-Santiago M, Duvauchelle CL, Crews D, Gore AC. 2018. Effects of the Endocrine-Disrupting Chemicals, Vinclozolin and Polychlorinated Biphenyls, on Physiological and Sociosexual Phenotypes in F2 Generation Sprague-Dawley Rats. Env Health Perspect https://doi.org/10.1289/EHP3550. Land PC, Owen EL, Linde HW. 1981. Morphologic changes in mouse spermatozoa after exposure to inhalational anesthetics during early spermatogenesis. Anesthesiol 54:53–6. Marczylo EL, Jacobs MN, Grant TW. 2016. Environmentally induced epigenetic toxicity: Potential public health concerns. Crit Rev Toxicol 46(8):676–700. Miska EA, Ferguson-Smith AC. 2016. Transgenerational inheritance: Models and mechanisms of non-DNA sequence-based inheritance. Science 354:6308;59–63. Nevison C, Blaxill M, Zaharodny W. 2018. California Autism Prevalence Trends from 1931 to 2014 and Comparison to National ASD Data from IDEA and ADDM. J Aut Dev Disord 48:12;4103–4117. Nilsson EE, Sadler-Riggleman I, Skinner MK. 2018. Environmentally induced epigenetic transgenerational inheritance of disease. Environ Epigenetics 4(2):dvy016. Nogueira FR, Braz LG, de Andrade LR, de Carvalho ALR, Vane LA, Modolo NSP, Aun AG, Souza KM, Braz JRC, Braz MG. 2016. Evaluation of Genotoxicity of General Anesthesia Maintained with Desflurane in Patients Under Minor Surgery. Env Mol Mutagen 57:312-316. Oropeza-Hernández LF, Quintanilla-Vega B, Albores A, Fernández-Guasti A. 2002. Inhibitory action of halothane on rat masculine sexual behavior and sperm motility. Pharmacol Biochem Behav 72:937–42. Pan JZ, Wei H, Hecker JG, Tobias JW, Eckenhoff RG, Eckenhoff MF. 2006. Rat brain DNA transcript profile of halothane and isoflurane exposure. Pharmacogenet Genomics 16:171–82. Pekny T, Andersson D, Wilhemsson U, Pekna M, Pekny M. 2014. Short general anaesthesia induces prolonged changes in gene expression in the mouse hippocampus. Acta Anaesthesiol Scand 58:9;1127-1133. Reiner O, Karzbrun E, Kshirsagar A, Kaibuchi K. 2015. Regulation of neuronal migration, an emerging topic in autism spectrum disorders. J Neurochem 136:3;440-456. Roustan A, Perrin J, Berthelot-Ricou A, Lopez E, Botta A, Courbiere B. 2012. Evaluating methods of mouse euthanasia on the oocyte quality: cervical dislocation versus isoflurane inhalation. Lab Anim 46:167–9. Schifilliti D, Mondello S, D’Arrigo G, Chille` G, Fodale V. 2011. Genotoxic effects of anesthetic agents: An update. Expert Opin Drug Saf 10:891–899. Tang CK, Chalon J, Markham JR, Ramanathan S.; Turndorf H. 1985. Exposure of Sires to Enflurane Affects Learning Function of Murine Progeny. Obstet Anesth Dig 5:2,67. Tang WWC, Dietmann S, Irie N, Leitch HG, Floros VI, Bradshaw CR, Hackett JA, Chinnery PF, Surani MA. 2015. A unique gene regulatory network resets the human germline epigenome for development. Cell 161(6):1453–1467. Tremblay MW, Jiang Y-H. 2019. DNA Methylation and Susceptibility to Autism Spectrum Disorder. Annu Rev Med 70:151–66. Wang LJ et al. 2008. Effects of inhaled anesthetics on human sperm motility in vitro. Brit J Anaesth 101:6;883-884. Western PS. 2018. Epigenomic drugs and the germline: Collateral damage in the home of heritability? Mol Cell Endocrin 468:121–133. Xu X, Pan C, Hu J, Liu X, Li Y, Wang H, Chen Y, Dong H,. Dai T, Xu L. 2012. Effects of isoflurane inhalation on the male reproductive system in rats. Env Toxicol Pharmacol 34:3:688-693. von Meyenn RW. 2015. Forget the parents: Epigenetic reprogramming in human germ cells. Cell 161:6;1248–1251. Vutskits L, Sall JW, Jevtovic-Todorovic V. 2018. A poisoned chalice: the heritage of parental anaesthesia exposure. Brit J Anesth 121:2;337-339. Yılmaz S, Çalbayram NC. 2016. Exposure to anesthetic gases among operating room personnel and risk of genotoxicity: A systematic review of the human biomonitoring studies. J Clinical Anesth 35:326-331.  View the videotaped presentations in this YouTube playlist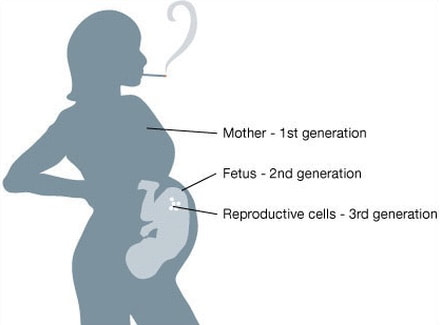 Smoking for three: pregnancy smoking can affect early germ cells developing within the embryo and fetus. Other periods of germline vulnerability will also be discussed at the workshop. Environmental Mutagenesis and Genomics Society Conference, Heritable Hazards of Smoking Workshop September 19, 2019, 8:45am to 3.30pm Washington, DC Abstract: Mounting evidence indicates that toxicant exposures to germ cells can impair the health and development of offspring in a variety of ways. Despite this, today’s regulatory paradigms focus on somatic impacts, with little consideration for the perturbation of the genetic and epigenetic features of germ cells or the heritable consequences thereof. This workshop will use the example of tobacco smoke as a case study to apply a modern next-generation testing paradigm, i.e., the Clean Sheet approach, in which we will encompass both somatic and germinal genomic damage in human risk assessment [Dearfield et al., Next Generation Testing Strategy for Assessment of Genomic Damage: A Conceptual Framework and Considerations. Environ Mol Mutagen 58:264-283, 2017]. The workshop consists of four parts: (1) background information about the Clean Sheet Initiative, tobacco toxicity, and germline vulnerabilities; (2) overview of evidence for germline impacts in humans and other mammals; (3) regulatory perspective; and (4) discussion geared toward consensus regarding needs for research and regulation. Webpage: https://www.emgs-us.org/p/cm/ld/fid=394 Registration: www.emgs-us.org/p/cm/ld/fid=386 Co-Chairs: Francesco Marchetti, Health Canada, Jill Escher, Escher Fund for Autism
Organizing Committee: Abigail Bline, UCLA, Kerry Dearfield, Retired (formerly at USDA and EPA), David DeMarini, US EPA, Jill Escher, Francesco Marchetti, Carole Yauk, Health Canada 8:45-9:00 AM Welcome, Opening Remarks, and Introductions Jill Escher, Escher Fund for Autism 9:00-9:20 AM The Clean Sheet Initiative and Its Potential to Identify the Risks of Heritable Consequences of Tobacco Smoke Exposures Kerry L. Dearfield, Retired (formerly at USDA and EPA), Burke, VA 9:20-9:40 AM Rationale for This Workshop: Growing Concern Regarding Heritable and Intergenerational Effects from Environmental Exposures Carole L. Yauk, Health Canada 9:40-10:00 AM The Potentially Vulnerable Periods of Exposure to the Male and Female Germline Jacquetta Trasler, McGill University, Montreal, ON, Canada 10:00-10:20 AM Coffee Break 10:20-10:40 AM Tobacco Smoke Exposure: Hazardous Components of Tobacco and Related products; Broad Overview of Known Health Effects; Tobacco-Induced Male Germ Cell and Heritable Effects Francesco Marchetti, Health Canada 10:40-11:00 AM Male-Mediated Heritable Epigenetic Effects Jacquetta Trasler, McGill University, Montreal, ON, Canada 11:00-11:20 AM Overview of Asthma and Allergy Epidemiology: Epigenetic Studies of Transgenerational Effects of Smoking John Holloway, University of Southhampton, UK 11:20-11:40 AM Multigenerational Transmission of Hyperactivity/ADHD Pradeep Bhide, Florida State University, Tallahassee, FL, USA 11:40-11:50 Grandmaternal Smoking and Risk for ADHD Gyeyoon Yim, ScD candidate, Harvard T.H. Chan School of Public Health, MA, USA 11:50-12:10 PM The RHINESSA Cohort, a Three-Generation Study on the Association of Tobacco Smoking with Asthma Cecillie Svanes, University of Bergen, Norway 12:10-12:30 PM Panel Discussion of the Evidence 12:30-1:30 PM Lunch (provided) 1:30-2:00 PM Toxicology of E-cigarettes Ilona Jaspers, University of North Carolina, NC, USA 2:00-2:30 PM Discussants Implications for Bioethics Anne Le Goff, Institute for Society and Genetics, UCLA, CA, USA Implications for Public Health Advocacy Laurent Huber, Executive Director, ASH, Washington, DC, USA 2:30-3:30 PM Directed Discussion and Consensus Opinion Methods and data to study intergenerational effects of these products and how to apply the Clean Sheet Framework. Is there a need for regulatory policy to emphasize germ cell risk in addition to somatic cell risk? Next steps? Research allows us to connect the molecular dots between germ cell exposure to GA and impaired brain development in offspring— and family histories reflect this pattern It is now generally accepted that toxicant exposures to germ cells during vulnerable stages of gametogenesis could "have a medically relevant effect on individual physiology" in offspring borne of the exposed cells (Perez et al., 2019). In particular, research in humans and mammals has repeatedly demonstrated that germ cell disruptions can result in dysregulated brain development and abnormal behaviors in offspring. Why does this occur? Multiple epigenetic factors including DNA methylation and histone modifications in the germ cells can be perturbed by exogenous toxicants, resulting to alterations of gene expression in the developing brain. (Reviewed in Gore et al., 2014; Walker et al., 2011; Yeshurun et al., 2018). Germ cells are at heightened sensitivity to epigenomic error during the early phases of gametogenesis (fetal and neonatal periods) when the DNA is globally demethylated and then remethylated in a sex-dependent manner. Abnormal patterning of DNA methylation or chromatin architecture in a particular region of germline in the developing fetus or neonate could lead to dysregulated somatic development in the generation borne of those exposed germ cells. This vulnerability of brain and behavior to germline perturbation has been observed with respect to a variety of toxicants, which I summarize here. Tobacco smoke, tobacco components, and related products can exert effects through exposed germ cells, resulting in abnormal brain function in offspring borne of exposed gametes. (McCarthy et al., 2018 [nicotine exposure in male mice produces behavioral impairment (hyperactivity, attention deficit, and cognitive inflexibility) in multiple generations of descendants]; Zhu et al., 2015 [grandpups of gestating mice exposed to nicotine exhibited behaviors comparable to attention deficit hyperactivity disorder (ADHD)]. Andalouss et al., 2018 [paternal exposure to cannabinoids during rat adolescence induces stress vulnerability in the offspring]; Golding et al., 2017 [human grandmaternal smoking linked to autism spectrum disorder (ASD) and autism trait risk in grandchildren through the female line]). Also Beal et al. performed a study to estimate the population effects of paternal smoking on prevalence of offspring intellectual disability, based on the occurrence of germline mutation (Beal et al. 2017). The same group found that tobacco smoke component benzo[a]pyrene increased levels of germline and somatic mosaicism in offspring, particularly in the brain. (Meier et al., 2017). Hormone-disrupting drug and chemical impacts on germ cells have been widely observed to disturb proper brain function in progeny. (Kioumourtzoglou et al., 2018 [in humans, significantly elevated odds for ADHD in the grandchildren of women who took diethylstilbestrol during pregnancy]; Moisiadis et al., 2017 [gestational treatment with betamethasone in guinea pigs at a clinically relevant dose resulted in various generational (through F3) pathology including altered cortisol response to stress, altered expression of genes in the prefrontal cortex and hypothalamic paraventricular nucleus]; Rawat et al,. 2018 [paternal corticosterone treatment in mice exerted effects on offspring brain serotonergic function]; Martinez et al., 2018 [exogenous thyroid hormone influences brain gene expression programs and behaviors in later generations by altering germ line epigenetic information]; Iqbal, et al., 2012 [gestational treatment with betamethasone modified HPA function and behavior in the F2 grandpup generation in guinea pigs borne of exposed germ cells]; Long et al., 2013 [dexamethasone administered in the clinical range to gestating ewes have multigenerational effects on HPA activity]; Krishnan et al., 2018 [exposure of rats to EDCs vinclozolin and polychlorinated biphenyls at the germ cell stage led to differences in the physiological and socio-sexual phenotype in offspring, especially in males]; Gillette et al., 2018 [gestational exposure to vinclozolin and PCBs in rats resulted in transgenerational inheritance of epimutations in brain and sperm]; Drobná et al. 2018 [transgenerational effects of BPA on gene expression and DNA methylation of imprinted genes in the mouse brain]; Crews et al. 2007 [females three generations removed from the original vinclozolin exposure discriminate and prefer males who do not have a history of exposure, in rats]; Crews et al., 2012 [a single exposure to vinclozolin altered the physiology, behavior, metabolic activity, and transcriptome in discrete brain nuclei in descendant male rats, causing them to respond differently to chronic restraint stress]; Wolstenholme et al, 2012 [gestational exposure to BPA produces multigenerational alterations in genes and behavior in mice]; Skinner et al., 2008 [gestating female rats were exposed to vinclozolin during fetal gonadal sex determination. Alterations to epigenetic reprogramming of the male germ-line and offspring brain transcriptome (sex-specific) were observed, Several brain signaling pathways were influenced including those involved in axon guidance and long-term potentiation]). General anesthetic gases have also been demonstrated to adversely impact brain and behavior of offspring borne of exposed germ cells. (Ju et al., 2018 [neonatal exposure to the widely used general anesthetic agent sevoflurane can affect the brains and behavior of the next generation of rat males through epigenetic modification of Kcc2 expression, while F1 females are at diminished risk]; Chalon et al., 1981 [learning retardation was seen in F2 mouse offspring of F1 parents exposed to general anesthesia in utero—in other words, mental impairment in the grandpups of the exposed gestating dams]; Tang et al., 1985 [general anesthetic agent enflurane administered to male mice was found to adversely affected learning function of their offspring]). Even opiates seem to exert intergenerational behavioral effects when delivered during sensitive stages of gametogenesis. (Vassoler et al., 2018 [morphine in F1 adolescent female rats, prior to conception, increases the rewarding effects of cocaine in F2 male and female offspring. Sex-specific alterations in endogenous opioids and hypothalamic physiology were observed]; Sabzevari et al., 2018 [morphine exposure to the F1 parent rat before conception induced intergenerational effects via dysregulation of HPA axis which results in anxiety in the adult male offspring]). This oft-demonstrated connection between germline disruption and brain-behavioral impairment in offspring—particularly with respect to tobacco smoke, synthetic steroid drugs, and general anesthesia—should sound the loudest of alarm bells throughout the research and public health spheres. Since the 1980s, the United States has experienced a staggering surge in the prevalence of idiopathic neurodevelopmental disorders we tend to label as ASD and ADHD. We know for certain these serious mental disabilities are highly heritable. But at the same time, we also know with reasonable certainty they are not highly genetic in any classic sense: “Genetic factors do not fully account for the relatively high heritability of neurodevelopmental conditions, suggesting that non-genetic heritable factors contribute to their etiology” (Martinez et al., 2018). It seems reasonable to ask whether the post-war boom in perinatal drugs, smoking and volatile synthetic anesthetic gases could have quietly perturbed the molecular integrity of early germ cells, resulting unexpectedly in a surge of neurodevelopmental abnormalities in the next generation. With this background in mind I'd like to discuss in particular the urgency of the question of heritable effects of general anesthesia. It has been known for some time that general anesthetics induce epigenetic modification of the genome. General anesthesia (GA) includes agents such as isoflurane, enflurane, halothane, and sevoflurane not only influence neuronal function, they also induce epigenetic alterations such a chromatin changes, histone modifications and shifts in DNA methylation (Csoka et al., 2009; Vutskits et al., 2018). GA agents can cause apoptosis and amyloid beta-protein accumulation, and neuronal damage, with potential mechanisms including enhanced protein misfolding and aggregation (Csoka et al., 2009). It has been shown that GA can cause substantial changes in gene and protein expression (Pan et al., 2006; Rampil et al., 2006). For example, even brief exposure to isoflurane leads to widespread changes in genetic control in the amygdala six hours after exposure (Pan et al., 2006). GA can modulate histone acetylation and as such may have deleterious effects on transcription of genes crucial for proper synapse formation and cognitive development (Dalla Massara et al., 2016). The above-cited study on sevoflurane demonstrated the induction of DNA methylation modification and changes in expression go brain-relevant genes. The investigators found a sex-specific decrease in KCC2 and increased DNA methylation of the KCC2 gene promoter in the sperm of F0 exposed sires (Ju et al., 2018). In an accompanying editorial, "A poisoned chalice: the heritage of parental anaesthesia exposure," Vutskits et al. noted that “we are faced with a real possibility that general anaesthetics are not innocuous agents that ‘only put children to sleep’ but rather formidable modulators of chromatin remodeling and function” (Vustskits et al, 2018). In spite of the ease with which one could connect the molecular dots from germ cell exposure to GA and impaired brain development in offspring, almost no research has been directed at this very critical question. Every day pregnant women and infants are treated with GA, and yet the potential deleterious effects on the fetal or early germ cells have been invisible to research and regulation. As someone deeply involved in the autism community, and who is in constant communication with autism families, I find this alarming, as I have observed certain patterns relating to germ cell exposure to GA/surgeries. The patterns I have observed seem to fall into three categories. Here, F0 = gestating mother of F1 and grandmother of F2; F1= parent of the autistic child; and F2 = autistic child. 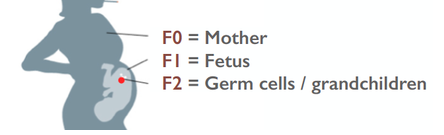 (1) F0 gestational exposure to GA. These are cases where the F0 grandmother of the F1 parent, male or female, had surgery during gestation with the F1. Reasons for the F0 surgeries during pregnancy included an appendectomy, surgery following an automobile accident, and surgery to correct a problem with the placenta. When an F1 parent had this prenatal exposure, he or she often had multiple F2 offspring, male and female, with autism. From a biological point of view this multiplex phenomenon would make sense because the early germ cells at this stage would likely be similarly exposed.
(2) F1 early childhood exposure to GA. These are cases where the F1 parent, male or female, had surgery or more typically a series of surgeries, in early life. Reasons for the F1 surgeries included tumor removal, hernia repair, surgeries to correct heart defects, and surgeries to correct birth defects such as clefts and club foot. When an F1 parent had this early life exposure, I saw he or she often had multiple F2 offspring, male and female, with autism. From a biological point of view this multiplex phenomenon might also make sense because the female oogonia are undergoing imprinting through the first year and are not yet mature, and the pre-meiotic male spermatogonial stem cells could also retain errors in their later-differentiated spermatocytes. (3) F1 paternal pre-puberty/puberty exposure to GA. These are cases where the F1 father had a series of surgeries around the time of puberty and beyond. In this category, two stories jump out. I have two male friends who suffered gunshot wounds in late childhood. Both underwent multiple surgeries in puberty and beyond to correct extensive damage. They each have one F2 son with extremely severe autism. Actually the word autism does not do their phenotypes justice, as their conditions are catastrophic, involving profound intellectual disability and severe behaviors, including in one case continuous and extreme self-injurious behaviors. In both of these cases the father also has F2 children who are typically developing. From a biological point of view this simplex phenomenon perhaps make sense because the germ cells were affected at a later stage of differentiation. It is worth nothing that in these cases, the families had no history of autism, and to my knowledge, the families and children had no risk factors for autism. As a rough control group, I noticed that where the F1 parent’s siblings did not have these sorts of surgical exposures, the F2s were typically developing. Given the molecular plausibility, the findings in mammal and human research literature, and the field observations discussed above, germline impact of general anesthesia is clearly an unexplored question of dramatic importance for public health that should be addressed without delay. —Jill Escher References Andalouss ZL, et al. Behavioural and epigenetic effects of paternal exposure to cannabinoids during adolescence on offspring vulnerability to stress. Int J Dev Neurosci 2019:72:48-54. Beal MA, et al. From sperm to offspring: Assessing the heritable genetic consequences of paternal smoking and potential public health impacts. Mut. Research/Rev in Mut. Research. 2017;773:26-50. Chalon J, et al. 1981. Exposure to halothane and enflurane affects learning function of murine progeny. Anesth. Analg.1981;60:794–7. Crews, D, et al. Transgenerational epigenetic imprints on mate preference. PNAS 2007;104 (14):5942-5946. Crews D, et al. Epigenetic transgenerational inheritance of altered stress responses. Proc Natl Acad Sci USA 2012;109:9143–8. Csoka AB, et al. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. Med. Hypoth. 2009;73:770–780. Dalla Massara L, et al. General anesthesia causes epigenetic histone modulation of c-fos and brain-derived neurotrophic factor, target genes important for neuronal development in the immature rat hippocampus. Anesthesiology 2016;124:1311e27. Drobná Z, et al. Transgenerational effects of bisphenol A on gene expression and DNA methylation of imprinted genes in brain. Endocrinology 2018;159:1132–144. Gillette R, et al. Passing experiences on to future generations: endocrine disruptors and transgenerational inheritance of epimutations in brain and sperm. Epigenetics 2018; https://doi.org/10.1080/15592294.2018.1543506. Golding J, et al. Grand-maternal smoking in pregnancy and grandchild’s autistic traits and diagnosed autism. Sci Rep 2017;7:46179. Gore AC, et al. Prenatal Steroid Perturbations for Neurodevelopment, Behavior, and Autism. Endocrine Rev 2014;35(6):961–991. Iqbal M, et al. Transgenerational effects of prenatal synthetic glucocorticoids on hypothalamic-pituitary-adrenal function. Endocrinol 2012;153, 3295–3307. Ju LS, et al., Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats. Brit. J Anesth 2018;121:2,406-416. Kioumourtzoglou M, et al. Association of Exposure to Diethylstilbestrol During Pregnancy With Multigenerational Neurodevelopmental Deficits. JAMA Pediatr 2018;172:7;670-677. Krishnan K, et al. Effects of the Endocrine-Disrupting Chemicals, Vinclozolin and Polychlorinated Biphenyls, on Physiological and Sociosexual Phenotypes in F2 Generation Sprague-Dawley Rats. Env. Health Perspect. 2018 https://doi.org/10.1289/EHP3550. Long NM, et al. Multigenerational effects of fetal dexamethasone exposure on the hypothalamic-pituitary-adrenal axis of first- and second-generation female offspring. Am J Obstet Gynecol 2013; 208, 217.e1–217.e8. Martinez ME, et al. Thyroid hormone influences brain gene expression programs and behaviors in later generations by altering germ line epigenetic information. Mol. Psych. 2018. https://www.nature.com/articles/s41380-018-0281-4 McCarthy, DM, et al. Nicotine exposure of male mice produces behavioral impairment in multiple generations of descendants. PLOS Biol, 2018;16(10):e2006497 DOI: 10.1371/journal.pbio.2006497. Meier MJ, et al. In utero exposure to benzo[a]pyrene increases mutation burden in the soma and sperm of adult mice. Environ Health Perspect 125:82–88; http://dx.doi.org/10.1289/EHP211 Moisiadis VG, et al. Prenatal Glucocorticoid Exposure Modifies Endocrine Function and Behaviour for 3 Generations Following Maternal and Paternal Transmission. Sci Rep 2017;7:11814. Pan JZ, et al. Rat brain DNA transcript profile of halothane and isoflurane exposure. Pharmacogenet Genomics 2006;16:171–82. Perez MF, et al. Intergenerational and transgenerational epigenetic inheritance in animals. Nat Cell Biol 2019. https://www.nature.com/articles/s41556-018-0242-9 Rampil IJ, et al. Isoflurane modulates genomic expression in rat amygdala. Anesth Analg 2006;102:1431–8. Rawat A, et al. Hypersensitivity to sertraline in the absence of hippocampal 5-HT1AR and 5-HTT gene expression changes following paternal corticosterone treatment. Env Epigenetics 2018;4(2):doi.org/10.1093/eep/dvy015. Sabzevari S, et al., Morphine exposure before conception affects anxiety-like behavior and CRF level (in the CSF and plasma) in the adult male offspring. Brain Research Bulletin 2019;144:122-131. Skinner MK, et al. Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS One 2008;3:e3745. Tang CK, et al. Exposure of sires to enflurane affects learning function of murine progeny. Obstet. Anesth. Dig. 1985;5:2,67. Vassoler F, et al., Increased cocaine reward in offspring of females exposed to morphine during adolescence. Psychopharmacology 2018;1–12. Vutskits L, et al. A poisoned chalice: the heritage of parental anaesthesia exposure. Brit. J Anesth. 2018;121;2,337-339. Walker, DM, et al. Transgenerational neuroendocrine disruption of reproduction. Nat Rev Endocrinol 2011;7: 197–207. Wolstenholme JT, et al. Gestational exposure to bisphenol A produces transgenerational changes in behaviors and gene expression. Endocrinology 2012;153:3828–38. Yeshurun S, et al. Transgenerational epigenetic influences of paternal environmental exposures on brain function and predisposition to psychiatric disorders. Mol Psychiatry 2018; doi: 10.1038/s41380-018-0039-z Zhu J, et al. Transgenerational transmission of hyperactivity in a mouse model of ADHD. J Neurosci 2014;34:2768–73.  The Escher Fund for Autism is pleased to announce its 2019 inaugural Escher Prizes in Germ Cell Exposures, recognizing researchers whose work shines a light on the heritable hazards of germline exposures to drugs or other chemicals. We invite you to nominate yourself or a colleague for one of these awards. Awardees will receive a $2,000 donation to help further their endeavors through their respective nonprofit institutions, as well as certificates of recognition.
Please help us recognize those who are breaking ground in revealing how germ cell exposure, particularly during the early stages of active reprogramming, to exogenous toxicants plays a non-genetic heritable role in the etiology of certain diseases and disorders, including but not limited to neurodevelopmental disorders, metabolic disorders, reproductive and socio-sexual disorders, asthma, allergies, and cancer. Areas of research may include, but are not limited to: epidemiology, animal models, in vitro assays, genomics, epigenomics and chromatin, toxicology, reproductive biology, chemical and pharmaceutical history, medical anthropology, and public health. Deadline to submit a nomination is March 31, 2019. Depending on the quality of the submissions, 2 to 4 prizes will be awarded. To submit a nomination, simply email us: (1) name, email address and institutional affiliation of the individual making the nomination; (2) name, email address and institutional affiliation of the nominee; and (3) up to 500 words explaining the reasons you believe the nominee deserves an award, providing citations to any relevant papers. Questions are welcome and may be directed here. Thank you, Escher Fund for Autism National Toxicology Program Office of Health Assessment and Translation (OHAT) Division of National Toxicology Program (NTP) National Institute of Environmental Health Sciences US Department of Health and Human Services Research Triangle Park, NC Submitted via NTP website December 11, 2018 Re: Nomination for scoping review of human and mammal evidence for adverse heritable impacts of direct germ cell exposure to drugs and chemicals Dear OHAT: In June of 2018 OHAT published a scoping review, “Human and animal evidence of potential transgenerational inheritance of health effects: An evidence map and state-of-the-science evaluation” (Walker et al., 2018). The immensely wide scope of the transgenerational report, the difficulty searching the literature, and the complexity of summarizing such a wide body of evidence—looking at different exposures, in different species, in different sexes, at different windows of developmental vulnerability, and with a plethora of different endpoints—surely made for a monumental undertaking. The undertaking seemed particularly difficult with regard to human exposures, considering that in most cohorts under study, insufficient time has passed to identify both F0 exposure and F3 outcomes. But more important, jumping to potential transgenerational effects without first ascertaining the state of the science on intergenerational inheritance induced by direct exogenous exposures to germ cells seemed hasty and premature—why concentrate on unexposed progeny while entirely skipping over the directly exposed (as a germ cell) offspring? This was a curious choice, especially in light of the fact that the American system of chemical and drug regulation currently ignores nearly all questions relating to direct germ cell impacts, creating a glaring need for scientific attention to this notably vulnerable window. This nomination asks OHAT to fill the void w ith a new scoping review devoted to questions of heritable (direct germ cell) effects of drugs and chemicals in human and mammal studies. Rationale for scoping review The second half of the 20th century witnessed a tsunami of novel chemical and pharmaceutical compounds, with countless newly devised substances entering the human body—and its germ cells—for the first time in the history of man. Beginning in the 1950s, scientists expressed alarm about the potential heritable force of some of these chemicals, leading to new approaches in toxicology as well as the founding of the Environmental Mutagenesis and Genomics Society (EMGS) in 1969 (Frickel, 2004; Wassom et al., 2010). Building on the original fears of the EMGS founders, the 21st century has seen a dramatic expansion of notions of inheritance to encompass molecular perturbation of germline by exogenous substances. While classic germ cell mutagenesis remains a stark concern in some cases (DeMarini, 2012), it is increasingly clear that exposures can induce adverse heritable effects quite apart from shifts in the DNA sequence—through the environmentally malleable layers of the epigenome of the germ cells (reviewed in Kexin et al. 2018; Skvortsova et al., 2018; Bonduriansky et al., 2018; Nilsson et al., 2018; Gapp et al., 2018; De Felici, et al., 2018; Marczylo et al., 2016). Indeed, a review of the scientific record reveals more than 100 studies in humans and mammals demonstrating adverse heritable effects of direct germ cell exposures (Escher, 2018b). None of this evidence of direct impact—which carries profound implications for understanding today’s public health challenges—was contemplated or included in the OHAT transgenerational scoping review of June of 2018. The American populace has been experiencing unprecedented rates of many serious pathologies, most of which cannot be explained by classic genetic or somatic environment paradigms. For example, the prevalence of autism, a serious lifelong neurodevelopmental disorder which entails vast societal cost, has surged to the point that 1 in 59 children are now identified as having autism spectrum disorders (Baio et al., 2018), with a prevalence observed to increase markedly with births in the 1980s (Nevison et al., 2018). Attention deficit hyperactivity disorder (ADHD) now affects 6.1 million children ages of 4 to 17, up from 4.4 million in 2003 (CDC, 2016). Obesity rates have surged, from a rare condition decades ago to 39.6% of adults in 2015-16 (Hales et al., 2018). Similarly, diabetes rates have surged dramatically (CDC, 2017). Sperm concentration and count have been dropping (Levine et al., 2017). The prevalence of polycystic ovary syndrome is as high as 15%–20% of women of childbearing age (Sirmans et al., 2014). The incidence of hypospadias has mysteriously doubled since the 1970s (Paulozzi et al., 1997). This is just a sample; many other conditions have been observed to be increasing in prevalence, with conventional “genetic” or “environment” approaches often unable to explain the numbers. Non-genetic inheritance caused by direct toxicant contamination of the germline may be a biological force driving up these mysteriously rising rates. If we apply contemporary understanding of germ cell toxicant vulnerabilities to pregnancy drug and chemical exposures of the postwar decades, one may reasonably ask whether today’s pathologies may be linked in part to those early, if forgotten, molecular interactions. For example, did maternal smoking, which reached its height in the 1960s, increase the risk for asthma, allergies, ADHD, and autism in grandchildren via exposure to the parents’ nascent germ cells? Several studies in mammals and humans suggest this may be true (Zhu et al., 2014; Rehan et al., 2013; Maritz et al., 2014, Golding et al., 2017; Accordini et al., 2018; Magnus et al., 2015; Lodge et al., 2018). Did synthetic steroid hormone drug exposure to fetal germ cells increase the risk for urogenital or neurodevelopmental abnormalities in the grandchild generation borne of the contaminated cells? Studies on diethylstilbestrol, for example, suggest this could be the case (Kioumourtzoglou et al., 2018; Tournaire et al., 2016). Does exposure to potent agents of general anesthesia damage primordial germ cells in a way that raises risks for learning and behavioral pathologies in the next generation offspring? Three published studies have demonstrated learning and behavioral impairment in the generation exposed as germ cells (Ju et al., 2018; Chalon et al., 1981; Tang et al., 1985). And countless other questions loom large. For example, did DDT and other pesticides quietly raise the risk for metabolic abnormalities in a generation borne of exposed germ cells? What about dioxin, chemotherapeutic agents, valproic acid, to name just a few? The evidence in the record thus far suggests these intergenerational questions are of tremendous importance to a full understanding of pathogenesis caused by exogenous toxicants. Moreover, the questions raised are not merely an exercise in retrospection. Every minute of every day, American children and adults ingest drugs and chemicals which may perturb the integrity of their germ cells, or, with respect to a pregnant woman, the integrity of her fetus’s germ cells as well. But risks to germ cells are routinely ignored in research, regulation, and practice. For example, if a man receives cortisone injections, there is no warning that the quality of his sperm may be temporarily affected. If a pregnant woman receives weekly injections of potent synthetic progestins, no one considers the potential impact on the fetal germline. If a woman is smoking tobacco, she is told of risks to the fetus, but not to her future grandchildren. If a couple visits a genetic counselor about their future offspring, the counselor will not even think to ask if either of them had been exposed to tobacco, synthetic steroids, or general anesthesia in utero. The United States is mired in archaic thinking about heritability, focusing solely on nucleotide sequence, while almost systematically ignoring germline vulnerabilities to toxicants. An OHAT scoping report on this topic could help change the national dialogue about pathogenesis of disease and disorder. It would examine the state of the evidence for intergenerational inheritance associated with direct germline exposure to chemicals and drugs in humans and mammals. By assessing the evidence for this form of non-genetic inheritance, it would help identify key areas of concern for public health and make recommendations for further research. Exposures to include in the scoping review This nomination seeks a review of research on heritable germline impact of exogenous chemicals and drugs. While other stressors such as diet, starvation, stress, and trauma could be considered as background material, this nomination is limited to exogenous chemicals and drugs (pharmaceutical and recreational, including tobacco and alcohol) only. The germline literature already includes research on the following:
For a list of human and mammal studies identified to date by the undersigned, please see germlineexposures.org (Escher, 2018b). Gametogenesis windows of vulnerability; mechanisms of interest It is clear that germline molecular impacts of toxicant exposure are highly dependent on the gametogenic window and sex of the germ cell. This introduces complexity to the present nomination, since analyses would need to be segregated by both timing of exposure and the sex of the exposed germ cell. Further complicating matters, germ cell exposures often yield sex-specific phenotypic outcomes in the progeny (see, for example, Ju et al., 2018; Krishnan et al., 2018; Golding et al., 2017). Background biological information is warranted. The epigenome refers to a heritable layer of biochemical information that includes DNA methylation, histone modifications, and non-coding RNAs. This information goes through exquisite reprogramming during the fetal germline phase. Because of epigenomic resetting, the fetal germ cells are at heightened sensitivity to perturbation by exogenous toxicants, which can act directly or indirectly to effectuate alterations in DNA methylation, histone modification and/or ncRNA expression (Gold et al., 2018; Marczylo et al., 2016). Abnormal hormonal signals, which may differ from endogenous signaling in terms of molecular structure, half life, and potency, can block, hyperactivate or otherwise disrupt normal hormone receptor function, ultimately influencing epigenomic regulation of gene expression (Marczylo et al., 2016). Because of these phenomena, “[e]pigenetic marks generated within germ cells as a result of environmental influences throughout life can also shape future generations long before conception occurs” (Bale, 2015). Below is an overview of commonly referenced critical windows, by sex. Note: the gestating woman is called “F0,” her exposed fetus is called “F1,” and the grandchildren borne of exposed fetal germ cells are called “F2.”  Exposures to female germ cells 1. Exposure of female F1 fetal germ cells via gestating F0. The male or female F1’s directly exposed embryonic/fetal germ cells later yield male or female grandchildren, or F2. Because embryonic/fetal germ cells are highly dynamic featuring multiple levels of reprogramming, as discussed above, this stage is considered an important critical window for exposure effects. 2. Exposure to a neonate female germ cells. Because the delicate process of genomic imprinting continues in the female ovary well after birth and through the first year, the infancy of the female may also present a critical window for exposures, though the molecular impacts are likely to be different than those seen in the primordial germ cell. 3. Exposure to germ cells of an adult pre-conception, non-gestating female. Some studies consider stressors to an adult female in the period well before or shortly before conception. The undersigned does not see much evidence for this window as a critical period of vulnerability for the egg epigenome, but is open to the possibility. That said, a pre-conception exposure of interest need not occur just before conception to be biologically relevant. A drug or chemical can be slow to metabolize, or may lodge in the fatty tissues even for decades (think of DDT or dioxin, for example). So long-gone exposures may still directly impact the contents of the egg that goes through the final stages of meiosis just prior to ovulation. Exposures to male germ cells  1. Exposure of male F1 fetal germ cells via gestating F0. This was discussed above.
2. Exposure to a neonate male germ cells. Similar to (2), above, except that the male germ cells have completed the imprinting process before birth. 3. Exposure to male germ cells during slow-growth period male. The period before puberty, when spermatogonia transition to spermatocytes. 4. Exposure to an adult pre-conception male germ cells. This is the final phase of spermatogenesis, the approximately 72 days in humans when primary spermatocyte matures into sperm. In sum, this nomination asks OHAT to undertake a scoping review of the state of the evidence for adverse heritable impacts of direct drug and chemical exposures to germ cells, in various stages of gametogenesis, but with a focus on the early germ cells which exhibit exceptional epigenomic vulnerability to toxicants. Again, an informal attempt at a first phase of this research, listing more than 100 human and mammal studies demonstrating direct germline effects, along with many reviews of the subject, has already been completed, with results published at germlineexposures.org (Escher, 2018b). As for endpoints, the literature suggests that neurodevelopment, neurobehavior, socio-sexual behaviors, and sexual development are often distorted as the result of germline exposure. Other endpoints of concern include asthma and allergy and metabolic dysfunction. Fifty years ago, leading scientists raised alarm bells about a possible ‘genetic emergency’ caused by the post-war influx of synthetic chemicals and drugs, concerned about subtle impairments in human germ cells that could affect the developmental integrity of future generations (Frickel, 2004; Escher, 2018a). Now that we understand germ cells not merely as an enclosure for a protein-coding template, but as complicated, highly dynamic biological entities that contain many layers of heritable information that can reshape gene expression and ultimately, brain development and behavior, it is time for taxpayer-funded research to embrace this biological reality, ascertain the risks, and take reasonable measures to safeguard the public. Thank you for your kind consideration of this nomination. Very truly yours, Jill Escher Cc: Vickie Walker, NIEHS Jack Bishop, NIEHS (ret) Jerry Heindel, NIEHS (ret) Linda Birnbaum, NIEHS Richard Woychik, NIEHS Francis Collins, NIH Diana Bianchi, NICHD Joshua Gordon, NIMH David DeMarini, EPA References Accordini S, Calciano L, Johannessen A, Portas L, Benediktsdóttir B, Bertelsen RJ, Bråbäck L, Carsin AE, Dharmage SC, Dratva J, et al. 2018. A three-generation study on the association of tobacco smoking with asthma. Int. J. Epidemiology;dyy031, https://doi.org/10.1093/ije/dyy031). Baio, J, Wiggins, L, Christensen, DL, Maenner, MJ, Daniels, J, Warren, Z, Kurzius-Spencer, M, Zahorodny, W, Rosenberg, CR, White, T, et al. 2018. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ 67:6;1–23. Bale, T. Epigenetic and transgenerational reprogramming of brain development. 2015. Nat Rev Neurosci 16;332–344. Bonduriansky R, Day T. 2018. Extended Heredity. Princeton, NJ: Princeton University Press. Chalon J, Tang CK, Ramanathan S, Eisner M, Katz R, Turndorf H. 1981. Exposure to halothane and enflurane affects learning function of murine progeny. Anesth Analg 60:794–7. DeMarini, DM. 2012. Declaring the Existence of Human Germ-CellMutagens. Environ Mol Mutagen 53:166-172. Escher J. 2018a. Bugs in the program: can pregnancy drugs and smoking disturb molecular reprogramming of the fetal germline, increasing heritable risk for autism and neurodevelopmental disorders? Environ Epigen 4:2;dvy001. Escher, J. Germline Exposures, Human and mammal studies finding heritable effects of exposures. 2018b. http://www.germlineexposures.org/studies-of-interest.html. Frickel S. 2004. Chemical Consequences: Environmental Mutagens, Scientist Activism, and the Rise of Genetic Toxicology. New Brunswick, NJ: Rutgers University Press p 87. Gapp K, Bohacek J. 2018. Epigenetic germline inheritance in mammals: looking to the past to understand the future. Genes Brain Beh 17:3,e12407. Gold HB, Jung YH, Corces VG. 2018. Not just heads and tails: the complexity of the sperm epigenome. J Biol Chem 293:36;13815-13820. Golding J, Ellis G, Gregory S, Birmingham K, Iles-Caven Y, Rai D, Pembrey M.l. 2017. Grand-maternal smoking in pregnancy and grandchild’s autistic traits and diagnosed autism. Sci Rep 7:46179. Hales CM, Fryar CD, Carroll MD, Freedman DS, Ogden CL. Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007-2008 to 2015-2016. 2018 JAMA;319(16):1723-1725. Ju LS, Yang JJ, Morey TE, Gravenstein N, Seubert CN, Resnick JL, Zhang JQ, Martynyuk AE. 2018. Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats. Brit J Anesth 121:2;406-4168. Kexin Z, et al. Current Advance in Research of Gamete and Embryo-fetal Origins of Adult Diseases. Science China Life Sciences 2018 10.1007/s11427-018-9427-4. Kioumourtzoglou M, Coull BA, O’Reilly ÉJ, Ascherio A, Weisskopf MG. Association of Exposure to Diethylstilbestrol During Pregnancy With Multigenerational Neurodevelopmental Deficits. 2018 JAMA Pediatr 172:7;670-677. Krishnan K, Nitish Mittal N, Thompson LM, Rodriguez-Santiago M, Duvauchelle CL, Crews D, and Gore AC. Effects of the Endocrine-Disrupting Chemicals, Vinclozolin and Polychlorinated Biphenyls, on Physiological and Sociosexual Phenotypes in F2 Generation Sprague-Dawley Rats. 2018 Env. Health Perspect. https://doi.org/10.1289/EHP3550. Levine H, Jørgensen N, Anderson M-A, Mendiola J, Weksler-Derri D, Mindlis I, Pinotti R, Swan SH. Temporal trends in sperm count: a systematic review and meta-regression analysis. 2017 Hum Reprod Update 23:6;646–659. Lodge CJ, Bråbäck L, Lowe AJ, Dharmage SC, Olsson D3, Forsberg B. 2018. Grandmaternal smoking increases asthma risk in grandchildren: a nationwide Swedish cohort. Clin. Exp. Allergy 48;167–74. Magnus MC, Håberg SE, Karlstad Ø, Nafstad P, London SJ, Nystad W. 2015. Grandmother’s smoking when pregnant with the mother and asthma in the grandchild: the Norwegian Mother and Child Cohort Study. Thorax 70:237-243. Marczylo EL et al, Environmentally induced epigenetic toxicity: potential public health concerns. Crit. Rev. Toxicol. 2016;46:8,676-700. Maritz GS, Mutemwa M. 2014. The effect of grand maternal nicotine exposure during gestation and lactation on lung integrity of the F2 generation. Pediatric Pulmonol 49:1;67-75. Nevison C, Blaxill M, Zaharodny W. 2018. California Autism Prevalence Trends from 1931 to 2014 and Comparison to National ASD Data from IDEA and ADDM. J Aut Dev Disord 48:12;4103–4117. Nilsson, EE, et al. Environmentally induced epigenetic transgenerational inheritance of disease. Environ Epigenetics. 2018;4(2). Paulozzi LJ, Erickson JD, Jackson RJ. 1997. Hypospadias trends in two US surveillance systems. Pediatrics 100:5;831-4. Rehan V, Liu J, Naeem E, Tian J, Sakurai R, Kwong K, Akbari O, Torday JS. 2012. Perinatal nicotine exposure induces asthma in second generation offspring. BMC Med 10:129. Sirmans SM, Pate KA. 2014. Epidemiology, diagnosis, and management of polycystic ovary syndrome. Clin Epidemiol 6:1–13. Skvortsova K, et al. Functions and mechanisms of epigenetic inheritance in animals. Nat Rev Molec Cell Biol 2018;19:774–790. Tang C-K, Chalon J, Markham JR, Ramanathan S.; Turndorf H. Exposure of Sires to Enflurane Affects Learning Function of Murine Progeny. Obstet Anesth Digest 1985;5:2,67. Tournaire M, Epelboin S, Devouche E, Viot G, Le Bidois J, Cabau A, Dunbavand A, Levadou A. 2016. Adverse health effects in children of women exposed in utero to diethylstilbestrol (DES). Therapie 71:4;395-404. U.S. Centers for Disease Control. 2016. Attention-Deficit / Hyperactivity Disorder (ADHD) https://www.cdc.gov/ncbddd/adhd/data.html (accessed November 9, 2018). U.S. Centers for Disease Control, Division of Diabetes Translation. 2017 Long-term Trends in Diabetes. https://www.cdc.gov/diabetes/statistics/slides/long_term_trends.pdf (accessed November 9, 2018). Walker VR, Boyles AL, Pelch KE, Holmgren, SD, Shapiro, AJ, Blystone, CR, Devito, MJ, Newbold RR, Blain R, Hartman P, et al. 2018. Human and animal evidence of potential transgenerational inheritance of health effects: An evidence map and state-of-the-science evaluation. Environ International 115:48-69. Wassom JS, Malling HV, Sankaranarayanan K, Lu P-Y. 2010. Reflections on the origins and evolution of genetic toxicology and the environmental mutagen society. 2010 Environ Mol Mutagen 51:8-9;746-760. Zhu J, Lee KP, Spencer TJ, Biederman J, Bhide PG. 2014. Transgenerational Transmission of Hyperactivity in a Mouse Model of ADHD. J Neurosci 34:8;2768-2773. National Toxicology Program
National Institute of Environmental Health Sciences US Department of Health and Human Services November 29, 2018 Re: Nomination for research on the heritable (germ cell) effects of general anesthetic agents Dear NTP: Each year in the United States 1.5 million young children and an unknown number of fetuses are acutely exposed to important and necessary, yet neurotoxic and epigenetically potent chemicals—general anesthetic gases (GA). While there has been ample research on somatic effects of these agents, there has been little attention to germ cell, or heritable, effects of exposure during critical windows of germline development. This represents a shocking void in the research, given that it is established that GA can cause substantial changes in gene and protein expression, and that animal models have consistently demonstrated intergenerational behavioral and learning impairment. Furthermore, in my experience as a leader in the autism community and as a research philanthropist, I have detected alarming patterns of neurodevelopmental pathology in offspring of parents who were heavily exposed to GA in very early life, during vulnerable periods of gametogenesis. (1) GA causes epigenetic modification of the genome It has been known for some time that general anesthetics induce epigenetic modification of the genome. General anesthesia includes agents such as isoflurane, enflurane, halothane, and sevoflurane not only influence neuronal function, they also induce epigenetic alterations such a chromatin changes, histone modifications and shifts in DNA methylation. (Csoka et al., 2009; Vutskits et al., 2018). GA agents can cause apoptosis and amyloid beta-protein accumulation, and neuronal damage, with potential mechanisms including enhanced protein misfolding and aggregation (Csoka et al., 2009). It has been shown that GA can cause substantial changes in gene and protein expression (Pan et al., 2009; Rampil et al., 2006). For example, even brief exposure to isoflurane leads to widespread changes in genetic control in the amygdala six hours after exposure (Pan et al., 2006). GA can modulate histone acetylation and as such may have deleterious effects on transcription of genes crucial for proper synapse formation and cognitive development (Dalla Massara et al., 2016). A recent study on sevoflurane demonstrated the induction of DNA methylation modification and changes in expression go brain-relevant genes. The investigators found a sex-specific decrease in KCC2 and increased DNA methylation of the KCC2 gene promoter in the sperm of F0 exposed sires (Ju et al., 2018). Germ cells are at heightened sensitivity to epigenomic error during the early phases of gametogenesis (fetal and neonatal periods) when the DNA is globally demethylated and then remethylated in a sex-dependent manner. Abnormal patterning of DNA methylation or chromatin architecture in a particular region of germline in the developing fetus or neonate could lead to dysregulated somatic development in the generation borne of those exposed germ cells. This is not mere speculation. Empirical evidence supports this idea. (2) Mammal models demonstrate intergenerational toxicity [Note F2 = autistic child; F1= parent; F0 = gestating mother of F1 and grandmother of F2.] The three mammal studies published on the question all point in the same direction. Most recently, neonatal exposure to the widely used general anesthetic agent sevoflurane (sub-clinical doses, in this case) affected the brains and behavior of the next generation of male rats through epigenetic modification of gene expression, while F1 females were at diminished risk (Ju LS, et al. 2018). The implications of this finding could be significant. To quote an accompanying editorial, ““Hence, we are faced with a real possibility that general anaesthetics are not innocuous agents that ‘only put children to sleep’ but rather formidable modulators of chromatin remodeling and function…. The current study extends previous reports of sex differences by showing that anaesthetic exposure itself can alter expression of chloride channels in certain brain regions and that this effect is heritable from exposed male parents to unexposed offspring” (Vutskits et al., 2018). The two earlier studies to observe offspring impairment are from the 1980s, well before epigenomic or chromatin mechanisms were under consideration. Learning retardation was seen in F2 mouse offspring of F1 parents exposed to general anesthesia in utero—in other words, mental impairment in the grandpups of the exposed gestating dams (Chalon et al., 1981). This phenomenon was also seen with the sires were exposed in adulthood. The general anesthetic agent enflurane administered to male mice was found to adversely affected learning function of their offspring (Tang et al., 1985). (3) Autism family case studies suggest GA acts as a potent germline toxicant during sensitive periods of gametogenesis For several years I have been discussing the question of parental fetal or early life exposures with autism parents. After numerous interviews I perceived a strong pattern of F2 pathology where the F1 had an early life exposure to GA, often by virtue of repeated and complicated surgeries. (a) F0 gestational exposure. These are cases where the F0 grandmother of the F1 parent, male or female, had surgery during gestation with the F1. Reasons for the F0 surgeries during pregnancy included an appendectomy, surgery following an automobile accident, and surgery to correct a problem with the placenta. When an F1 parent had this prenatal exposure, he or she often had multiple F2 offspring, male and female, with autism. From a biological point of view this multiplex phenomenon would make sense because the early germ cells at this stage would likely be similarly exposed. (b) F1 early childhood exposure. These are cases where the F1 parent, male or female, had surgery or a series of surgeries, in early life. Reasons for the F1 surgeries included tumor removal, hernia repair, surgeries to correct heart defects, and surgeries to correct birth defects such as clefts and club foot. When an F1 parent had this early life exposure, I saw he or she often had multiple F2 offspring, male and female, with autism. From a biological point of view this multiplex phenomenon might also make sense because the female oogonia are undergoing imprinting through the first year and are not yet mature, and the pre-meiotic male spermatogonial stem cells could also retain errors in their later-differentiated spermatocytes. (c) F1 paternal pre-puberty/puberty exposure. These are cases where the F1 father had a series of surgeries around the time of puberty and beyond. In this category, two stories jump out. I have two male friends who suffered gunshot wounds in late childhood. Both underwent multiple surgeries in puberty and beyond to correct extensive damage. They each have one F2 son with extremely severe autism. Actually the word autism does not do their phenotypes justice, as their conditions are catastrophic, involving profound intellectual disability and severe behaviors, including in one case continuous and extreme self-injurious behaviors. In both of these cases the father also has F2 children who are typically developing. From a biological point of view this simplex phenomenon perhaps make sense because the germ cells were affected at a later stage of differentiation. I am aware these are all anecdotes and alone prove nothing. But it is worth nothing that in these cases, the families had no history of autism, and to my knowledge, the families and children had no risk factors for autism. As a rough control group, I noticed that where the F1 parent’s siblings did not have these sorts of surgical exposures, the F2s were typically developing. In spite of the importance of the questions presented, the NTP might respond, “we only study environmental chemicals, and GA falls under the purview of the FDA, not the NIEHS.” In spite of that, I entreat the NTP to include these exposures in the NTP for the following reasons:
The question remains: which agents to study? Because halothane was the most popular GA during the post-war decades and sevoflurane is popular today, I would suggest the NTP begin with those two agents and then expand to others depending on the signals from pilot data. It may turn out that thorough investigation of germline effects (via many different windows of germline development, many different doses, and/or many different mixtures) may well be warranted. Thank you for your kind consideration of this nomination. Very truly yours, Jill Escher References Chalon J, Tang CK, Ramanathan S, Eisner M, Katz R, Turndorf H. 1981. Exposure to halothane and enflurane affects learning function of murine progeny. Anesth Analg 60:794–7. Csoka AB, Szyf M. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. 2009. Med. Hypoth. 73;770–780. Dalla Massara L, Osuru HP, Oklopcic A, et al. General anesthesia causes epigenetic histone modulation of c-fos and brain-derived neurotrophic factor, target genes important for neuronal development in the immature rat hippocampus. 2016. Anesthesiology 124:1311e27. Heflich R. 2018. Lecture at Environmental Mutagenesis and Genomics 2018 Meeting, “My Forty Years at the U.S. Food and Drug Administration, Robert H. Heflich, U.S. Food and Drug Administration.” Delivered September 24, 2018. Ju LS, Yang JJ, Morey TE, Gravenstein N, Seubert CN, Resnick JL, Zhang JQ, Martynyuk AE. 2018. Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats. Brit J Anesth 121:2;406-4168. Martinez et al., Thyroid hormone influences brain gene expression programs and behaviors in later generations by altering germ line epigenetic information, Mol. Psych. 2018. Pan JZ, Wei H, Hecker JG, Tobias JW, Eckenhoff RG, Eckenhoff MF. Rat brain DNA transcript profile of halothane and isoflurane exposure. Pharmacogenet Genomics 2006;16:171–82. Rampil IJ, Moller DH, Bell AH. Isoflurane modulates genomic expression in rat amygdala. Anesth Analg 2006;102:1431–8. Tang CK, Chalon J, Markham JP, Ramanathan S, Turndorf H. Exposure of sires to enflurane affects learning function of murine progeny. 1985. Obstet. Anesth. Dig. 5:2,67. Vutskits L, Sall JW, Jevtovic-Todorovic V. A poisoned chalice: the heritage of parental anaesthesia exposure. 2018. Brit. J. Anesth. 121;2,337-339. Woodcock, J. Letter to Escher in response to citizens petition to withdraw approval for 17-alpha hydroxyprogesterone caproate as a drug used in pregnancy. 2018. http://www.germlineexposures.org/uploads/6/4/0/9/6409433/fda-2015-p-0876.pdf (accessed November 9, 2018). [This rehashes a previous blog entry from 2014.] By Jill Escher How pervasive was pregnancy drug use in the 1960s? We can look at two cohorts, the CHDS (Child Health and Development Studies) and the CPP, or Collaborative Perinatal Project to get a sense of this vast history. The top chart shows CHDS drug use throughout pregnancy, and the bottom two refer to drug CHDS and CPP drug use in roughly the first half of pregnancy. Use of anti-nausea drugs, sedatives, hormones, painkillers and amphetamines were all common. Which of these abnormal exposures entered the womb and fetal tissue? All of them. Which of these affected fetal development? Most of them, though sometimes very subtly. Which of them affected fetal germline? Well, we hope to find out. 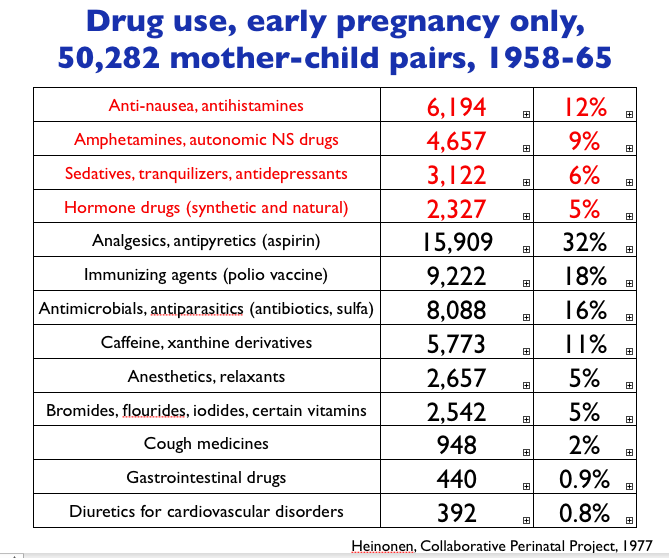 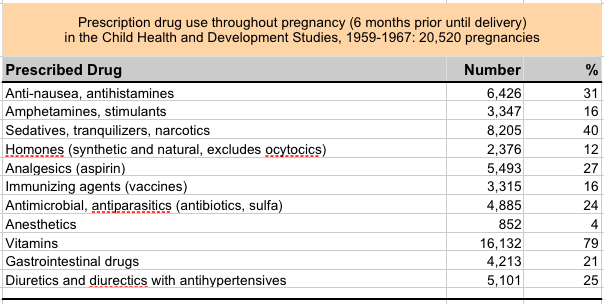 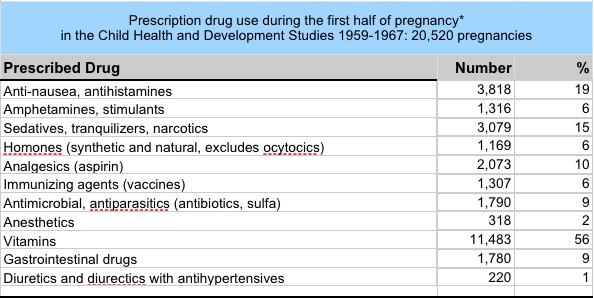 Escher Fund for Autism
1590 Calaveras Avenue San Jose, CA 95126 Francis Collins, Director, National Institutes of Health Diana Bianchi, Director, National Institute of Child Health and Human Development Joshua Gordon, Director, National Institute of Mental Health Linda Birnbaum, Director, National Institute of Environmental Health Sciences Via Email and U.S. Mail November 4, 2018 Re: Request for NIH research program on the heritable effects of general anesthesia Dear Drs. Collins, Bianchi, Gordon and Birnbaum: Let me start this missive by acknowledging that you probably receive a number of letters from community members opining about potential sources of the increasing prevalence of autism and related disorders. Now in my seventh year of funding pilot studies relating to autism, I have grown to share the research community’s skepticism toward the out-of-the-box ideas regarding causation, because, well, most of those ideas are just plain wrong. However, I am writing today about a very particular concept that has leapt out at me as deserving serious attention at the NIH: the question of heritable, germline-borne effects of agents of general anesthesia. My work as a funder of pilot studies in non-genetic inheritance began nearly seven years ago shortly after I discovered I had been very heavily and continuously exposed in utero to powerful synthetic steroid hormone drugs, prompting the idea that hormone signal disruption to my fetal primordial germ cells could have perturbed the epigenomic programming of my eggs, thus causing dysregulation of gene expression, seemingly limited to the prefrontal cortex, in my two children who have idiopathic nonverbal autism [Escher J, Bugs in the Program: can pregnancy drugs and smoking disturb molecular reprogramming of the fetal germline, increasing heritable risk for autism and neurodevelopmental disorders? Environ. Epigen. 2018;4:2]. To further develop hypotheses regarding non-genetic inheritance in neurodevelopmental disorders, I delved into the scientific literature regarding direct exposures to germline, engaged deeply with the scientific community, and interviewed autism families in an effort to detect patterns regarding parental fetal or early life exposures and adverse outcomes in their children. A few years ago, I noticed a number of stray comments (as I was not asking about surgeries at all at first) that emerged as a strong pattern. Many of the parents made remarks about early exposure to surgery. These stories seemed to fall into three categories. [Here F2 = autistic child; F1= parent; F0 = gestating mother of F1 and grandmother of F2.] (1) F0 gestational exposure. These are cases where the F0 grandmother of the F1 parent, male or female, had surgery during gestation with the F1. Reasons for the F0 surgeries during pregnancy included an appendectomy, surgery following an automobile accident, and surgery to correct a problem with the placenta. When an F1 parent had this prenatal exposure, he or she often had multiple F2 offspring, male and female, with autism. From a biological point of view this multiplex phenomenon would make sense because the early germ cells at this stage would likely be similarly exposed. (2) F1 early childhood exposure. These are cases where the F1 parent, male or female, had surgery or a series of surgeries, in early life. Reasons for the F1 surgeries included tumor removal, hernia repair, surgeries to correct heart defects, and surgeries to correct birth defects such as clefts and club foot. When an F1 parent had this early life exposure, I saw he or she often had multiple F2 offspring, male and female, with autism. From a biological point of view this multiplex phenomenon might also make sense because the female oogonia are undergoing imprinting through the first year and are not yet mature, and the pre-meiotic male spermatogonial stem cells could also retain errors in their later-differentiated spermatocytes. (3) F1 paternal pre-puberty/puberty exposure. These are cases where the F1 father had a series of surgeries around the time of puberty and beyond. In this category, two stories jump out. I have two male friends who suffered gunshot wounds in late childhood. Both underwent multiple surgeries in puberty and beyond to correct extensive damage. They each have one F2 son with extremely severe autism. Actually, the word autism does not do their phenotypes justice, as their conditions are catastrophic, involving profound intellectual disability and severe behaviors, including in one case continuous and extreme self-injurious behaviors. In both of these cases the father also has F2 children who are typically developing. From a biological point of view, this simplex phenomenon perhaps make sense because the germ cells were affected at a later stage of differentiation. Of course these are all anecdotes, and alone prove nothing. But it is worth noting that in these cases, the families had no history of autism, and to my knowledge, the families and children had no risk factors for autism. As a rough control group, I noticed that where the F1 parent’s siblings did not have these sorts of surgical exposures, the F2s were typically developing. More important, though, is the fact that animal models of heritable effects of general anesthesia exposures demonstrate a biological plausibility. Animal models demonstrate biological plausibility While there is surprisingly little literature on heritable effects of general anesthesia—considering that many of the agents commonly used in the 1950s through today are known to be genotoxic and epigenetically disruptive—the three mammal studies published on the question all point in the same direction. Most recently, neonatal exposure to the widely used general anesthetic agent sevoflurane (sub-clinical doses, in this case) affected the brains and behavior of the next generation of male rats through epigenetic modification of gene expression, while F1 females were at diminished risk [Ju LS, et al., Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats. Brit. J Anesth. 2018;121:2,406-416]. The implications of this finding could be significant. To quote an accompanying editorial, ““Hence, we are faced with a real possibility that general anaesthetics are not innocuous agents that ‘only put children to sleep’ but rather formidable modulators of chromatin remodeling and function…. The current study extends previous reports of sex differences by showing that anaesthetic exposure itself can alter expression of chloride channels in certain brain regions and that this effect is heritable from exposed male parents to unexposed offspring.” [Vutskits L, et al. A poisoned chalice: the heritage of parental anaesthesia exposure. Brit. J Anesth. 2018;121;2,337-339.] The two earlier studies to observe offspring impairment are from the 1980s, well before epigenomic or chromatin mechanisms were under consideration. Learning retardation was seen in F2 mouse offspring of F1 parents exposed to general anesthesia in utero—in other words, mental impairment in the grandpups of the exposed gestating dams [Chalon J, et al. Exposure to halothane and enflurane affects learning function of murine progeny. Anesth. Analg.1981;60:794–7]. This phenomenon was also seen with the sires were exposed in adulthood. The general anesthetic agent enflurane administered to male mice was found to adversely affected learning function of their offspring [Tang CK, et al. Exposure of sires to enflurane affects learning function of murine progeny. Obstet. Anesth. Dig. 1985;5:2,67]. When I attended the Environmental Mutagenesis and Genomics Society meeting in September of this year, several scientists, agreeing about the need for a research program regarding these questions, suggested I contact the NIEHS NTP {National Toxicology Project]. When I attended the Cold Spring Harbor Laboratories meeting on Germ Cells in October of this year, several of the scientists also saw the value of these questions, and recommended that I suggest a research program to the NICHD. However, with regard to the FDA’s National Center for Toxicological Research, my discussions with Robert Heflich of the NCTR were not promising, as his department appeared to be neither interested in germ cells nor epigenomic disruptions. Accordingly, I write NIH leaders to ask, what will it take to create a research program specifically devoted to these questions of non-genetic inheritance caused by germ cell exposure to agents of general anesthesia in use from the 1950s to today? While I do plan to submit a nomination to the NIEHS NTP on this topic, I feel that that, while possibly a useful avenue, that is alone insufficient to address the great breadth and weight of these questions. Since the 1980s, the United States has experienced a staggering surge in the prevalence of idiopathic neurodevelopmental disorders we tend to label as autism. We know for certain these serious mental disabilities are highly heritable. We also know with reasonable certainty they are not highly genetic in any classic sense [“Genetic factors do not fully account for the relatively high heritability of neurodevelopmental conditions, suggesting that non-genetic heritable factors contribute to their etiology.” Martinez et al., Thyroid hormone influences brain gene expression programs and behaviors in later generations by altering germ line epigenetic information, Mol. Psych. 2018.] In light of that and the foregoing it makes sense to shift some attention to routes of non-genetic inheritance. While I believe that a great many ancestral germline exposures should concern us (my principal focus remains pregnancy drugs), I cannot help but see the germline effects of general anesthesia issue as an unexplored question of dramatic importance for public health that alone should be addressed without delay. If the NIH or any of its branches would take an interest in what I propose. I have several ideas for research programs in both human cohorts and animal models, and would of course be enthusiastic to share them with you. I can be reached at [email protected]. Thank you for your kind consideration of these comments. Very truly yours, Jill Escher Cc via Email: Robert Heflich, FDA; Rosalie Elespuru, FDA; Sharvani Mahadevaraju, NIH; Daniel Camerini-Otero, NIH; Louis Reichardt, Simons Foundation; Thomas Frazier, Autism Speaks The idea of “conditions of life” affecting germ cells is so old-school it can be traced back to The Origin of Species in 1859  "I believe that the conditions of life, from their action on the reproductive system, are so far of the highest importance as causing variability,” wrote Darwin 159 years ago. By Jill Escher
The TwitterSphere has been crackling lately with heated debate about epigenetic inheritance—is it a Thing? Is it distracting Lamarckian, anti-Darwinian nonsense? Does it even matter for science or human health? Being firmly on the side of yaaaaaasss, intergenerational epigenetic inheritance is for sure a Thing, and really super important Thing for humans and other beasts at that, I got my share of virtual eye rolls for holding such a “radical” idea. In the opposite corner of the ring was Kevin Mitchell, who has made a name for himself as an entrenched skeptic of both intergenerational (involving a direct germline stressor) and transgenerational inheritance (involving no direct germ cell exposure). In recent blogs (here and here) he insisted the scientific literature was devoid of convincing evidence of even intergenerational epigenetic inheritance in mammals or humans (which I pointed out, citing an abundance of studies, was egregious overstatement, here). Shaken by his dogmatism and hyperbole, I shed a little tear and consoled myself in the pages of pretty good book, called The Origin of Species, by my one of favorite guys, Charles Darwin. Darwin’s theory of evolution has often been summed up as “random mutation and natural selection,” but anyone who has read Origin must be aware, Darwin’s views on the source of variation had little to do with randomness (or with mutations, as he was not remotely aware of DNA or genes). Darwin puzzled over the forces behind variation and admitted he lacked a clear answer, but he insisted he was “strongly inclined” toward the existence of a certain phenomenon, which he addressed multiple times in Origin. What phenomenon? Well, it’s something strikingly consistent with the idea of intergenerational epigenetic inheritance. Darwin introduces the concept in Chapter 1, Variation under Domestication. He writes that while variability may be induced by exposures to the embryo, “I am strongly inclined to suspect that the most frequent cause of variability may be attributed to the male and female reproductive elements having been affected prior to the act of conception.” (The Origin of Species, p. 10) In other words, Darwin thinks some experiences of the sperm and egg at some point(s) before conception, generate variation in how they are expressed. He then explains his chief reason for suspecting this is his observation in domestic animals that the reproductive system appears “to be far more susceptible than any other part of the organization, to the action of any change in the conditions of life.” (Id. 11) In plants he notes that “trifling changes, such as a little more or less water at some particular period of growth, will determine whether or not the plant sets a seed.” (Id.) He then explain that plant sports, which he sees as common under cultivation, support his view “that variability may be largely attributed to the ovules or pollen, or to both, having been affected by the treatment of the parent prior to the act of conception.” (Id. 12) He goes on to argue against direct (what we today would call prenatal somatic) exposures as having much influence over variability in animals, though he sees more such influence in plants. (Id. 12-13) Lest you think his musings on germline exposures (to use today’s language) generating variation in offspring are a fluke, please consider how often he returns to this theme. After stating that the continual breeding of horses, cattle and poultry for an “almost infinite number of generations” is obviously possible based on society’s long experience of domestication, he adds “that when under nature the conditions of life do change, variations and reversions of character probably do occur; but natural selection, as will hereafter be explained, will determine how far the new characters thus arising shall be preserved. (Id. 17) In other words, environmental context in nature can probably drive development of variations, on which natural selection then acts. Darwin closes Chapter 1 with a summary, reiterating, “I believe that the conditions of life, from their action on the reproductive system, are so far of the highest importance as causing variability,” then he admits “variability is governed by many unknown laws, with “the final result is thus rendered infinitely complex.” (Id. 44) Now let’s move on to Chapter 5, “Laws of Variation.” He starts off by dismissing the idea of “chance” as the force generating variation in offspring. He says “I have hitherto sometimes spoken as if the variations so common and multiform in organic beings under domestication, and in a lesser degree in those in a state of nature--had been due to chance. This, of course, is a wholly incorrect expression, but it serves to acknowledge plainly our ignorance of the cause of each particular variation.” (Id. 133) He asserts the greater force in creating variability, as well as “monstrosities” under domestication or cultivation, “are in some way due to the nature of the conditions of life, to which the parents and their more remote ancestors have been exposed during several generations.” Oh dear, that's transgenerational, or at least cumulative-generational, inheritance— this man was truly a hypothesis-maker way ahead of his time (hat tip here to Pat Hunt's recent study, Horan TS, et al. Germline and reproductive tract effects intensify in male mice with successive generations of estrogenic exposure. PLOS Genetics 2017;1006885). He repeats that the “reproductive system is eminently susceptible to changes in the conditions of life; and to this system being functionally disturbed in the parents, I chiefly attribute the varying or plastic condition of the offspring.” (Id. 133-34) I nearly fainted when I read that last sentence. With joy, with relief, with sheer worshipfulness of Mr. Darwin. This sums up my work, my hypothesis, my life story. While I won’t go into the details here you can read them in Bugs in the Program, published in the journal Environmental Epigenetics. In short, I contend my early “reproductive system’s” (germ cells) heavy prenatal exposure to a bizarre “condition of life” (huge doses of synthetic steroid hormone drugs) caused the “varying condition” of my offspring (dysregulated neurodevelopment). It appears this game of connect-the-dots would not have fazed Mr. Darwin at all. And, to further cheer me up, I guess, he then emphasizes it again: "The male and female sexual elements seem to be affected before that union takes place which is to form a new being…. we may feel sure that there must be some cause for each deviation of structure, however slight.” (Id. 134) Then, he again dismisses the idea of proximate somatic exposures having much effect. “How much direct effect difference of climate, food, &c., produces on any being is extremely doubtful.” The totality of traits results from some mix “accumulative action of natural selection” and “conditions of life.” (Id. 135) But he is pretty certain that “conditions of life" must be acting “indirectly,” by which he means “affecting the reproductive system, and in thus inducing variability.” After that, "natural selection will then accumulate all profitable variations, however slight, until they become plainly developed and appreciable by us.” (Idd 136) Toward the close of the book, Darwin reiterates the causes of variation act indirectly, through conditions affecting the parental reproductive elements. The cause of variation “I believe generally has acted, even before the embryo is formed; and the variation may be due to the male and female sexual elements having been affected by the conditions to which either parent, or their ancestors, have been exposed." (Id. 433-434) Darwin could not possibly have been more prescient when he wrote in his final pages, “A grand and almost untrodden field of inquiry will be opened, on the causes and laws of variation, on correlation, on the effects of use and disuse, on the direct action of external conditions, and so forth.” (Id. 474) For decades, however, science has been tragically stuck in the rut of “random mutation” as the cause of variation, and a rigid sort of genetic determinism, in clear contravention of Darwin’s own views. It's only now that this untrodden field is finally opening to its true breadth. One further note: Several years after publishing The Origin of Species, Darwin further hypothesized that tiny "gemmules" took information from the body to the germ cells, influencing traits of offspring. We now know of transfer of various forms of somatic molecular information to germ cells via cytoplasmic bridges. Two more points for Chuck. I hope my work can play a small part in the flowering of Darwin’s original ideas about exposures to the parental germ cells influencing traits in offspring. Today there is little doubt the germline epigenome, as influenced by exposures to the germ cells, can actively play a role in determining gene expression, and thus traits, in the resulting organism. To this, Darwin would not say "What a radical idea!" but rather, “Of course, I've been saying that for 159 years!” [All emphasis, mine.] Source: Darwin, Charles, 1809-1882. On The Origin of Species by Means of Natural Selection, or Preservation of Favoured Races in the Struggle for Life. Original text of first ed London:John Murray, 1859. In mass market reissue, New York:Bantam Classic, 2008. Jill Escher is a research philanthropist. Learn more at GermlineExposures.org. |
AuthorJill Escher, Escher Fund for Autism, is a California-based science philanthropist and mother of two children with severe autism, focused on the question of how environmentally induced germline disruptions may be contributing to today's epidemics of neurodevelopmental impairment. You can read about her discovery of her intensive prenatal exposure to synthetic hormone drugs here. Jill is also president of Autism Society San Francisco Bay Area. Archives
July 2021
Categories |

- Home
-
Expert Q&A
- Eva Jablonka Q&A
- Amander Clark Q&A
- Mirella Meyer-Ficca Q&A
- Janine LaSalle Q&A
- Dana Dolinoy Q&A
- Ben Laufer Q&A
- Tracy Bale Q&A
- Susan Murphy Q&A
- Alycia Halladay Q&A
- Wendy Chung Q&A
- Pradeep Bhide Q&A
- Pat Hunt Q&A
- Yauk and Marchetti Q&A
- Emilie Rissman Q&A
- Carol Kwiatkowski Q&A
- Linda Birnbaum Q&A
- Virender Rehan Q&A
- Carlos Guerrero-Bosagna Q&A
- Randy Jirtle Q&A
- Jerry Heindel Q&A
- Cheryl Walker Q&A
- Eileen McLaughlin Q&A
- Carmen Marsit Q&A
- Marisa Bartolomei Q&A
- Christopher Gregg Q&A
- Andrea Baccarelli Q&A
- David Moore Q&A
- Patrick Allard Q&A
- Catherine Dulac Q&A
- Lucas Argueso Q&A
- Toshi Shioda Q&A
- Miklos Toth Q&A
- Piroska Szabo Q&A
- Reinisch Q&A
- Klebanoff Q&A
- Denis Noble Q&A
- Germline in the News
- Science
- Presentations
- About Us
- Blog
 RSS Feed
RSS Feed
Proudly powered by Weebly