No Convincing Evidence? A Response to Kevin Mitchell’s Reckless Attack on Epigenetic Inheritance7/18/2018  By Jill Escher With no small amount of alarm I recently read Kevin Mitchell’s dismissal of the idea that environmentally informed nongenetic inheritance plays a role in the etiology of human traits and pathologies. I’m referring to his May 29, 2018 Wiring the Brain blogpost, “Grandma’s trauma – a critical appraisal of the evidence for transgenerational epigenetic inheritance in humans.” Now, I hardly mind critical appraisals of evidence in epigenetics or anything else, but the resounding sweep of his conclusions is so overstated and divorced from reality I felt I could not let it stand without comment. Mitchell, Associate Professor in Genetics and Neuroscience at Trinity College, Dublin, contends “there is no convincing evidence showing transgenerational epigenetic inheritance in humans.” His implication is clear: only “sociological reasons,” and not sound science, lie behind the continuing scientific and popular interest in this “sexy story,” which based on his listing of alleged sociological reasons seems to involve nothing more than an echo chamber of lazy researchers unnecessarily diverting attention from that King of Inheritance, determinism via the DNA sequence. There is nothing remotely subtle about Mitchell’s contempt for the science of epigenetic inheritance in mammals and humans. He states that the “idea that a person’s experiences can somehow mark their genomes in ways that are passed on to their children and grandchildren” then influencing “gene expression and affect[ing] the behaviour and physiology of people who inherit them” is “based on the flimsiest of evidence from a very small number of studies with very small sample sizes and serious methodological flaws.” Most alarmingly (at least to me), he scoffs at any implication of “epigenetics as a crucial new mechanism in medicine and public health – both a cause of disease and a potential therapeutic target.” While I won’t quibble with the “therapeutic target” part, the contention that epigenetic inheritance is unimportant for public health… well, as a prenatally-exposed-to-crud person myself, one who loudly champions the need for more research in this field (see “Bugs in the Program” in the journal Environmental Epigenetics), that hit me right in the gut. So I urge readers to reconsider his broadside. After all, scientific ideas have consequences. And since Mitchell’s post got a lot of TwitterLove, not to mention a stamp of approval from Jerry Coyne, I fear Mitchell’s words could, however subtly, dampen interest in an area I think deserves a rave-party level of enthusiasm. Much is at stake in this debate, including our hive-mind willingness to explore the heritable risks of pregnancy drugs, chemicals, smoking and trauma. "Much is at stake here, including our hive-mind willingness to explore the heritable risks of pregnancy drugs, chemicals, smoking and trauma." We pause for a moment of linguistic clarity There are many definitions floating around for “transgenerational,” “epigenetic” and related terms and phrases, and I hardly blame Mitchell for lexical mushiness seen in his and many other writings (including my own). But when he derides “transgenerational epigenetic inheritance,” I have difficulty figuring out what he actually means, from a biological point of view. “Transgenerational” usually refers to inherited traits without any direct exposure to the soma or germline. In contrast, “intergenerational” phenomena usually refer to heritable results of a direct exposure to the germline. Mitchell looks at studies investigating three cohorts: offspring and grand offspring of men and women living in Överkalix, Sweden (variable food supply); offspring of women gestating during the Dutch Famine (suffering often severe malnutrition); and grandoffspring of pregnant women in São Gonçalo, Brazil (exposed to violence). For the most part the phenomena at issue in these studies are not “transgenerational,” but rather “intergenerational.” So I’ll use that term. [Because these processes are clearly extremely sex- and developmental window- specific, a certain degree of biological precision is warranted. Appendix 1 provides a starter-kit outline for those who want to learn more.] Lamenting epigenetics hype in the popular media is cute, but irrelevant As an aside, and this hardly seems worth addressing, but Mitchell invokes media hype around epigenetics to heighten a dramatic contrast to supposedly “flimsy” findings in research. Sure, media hype happens. But his argument is like blaming researchers for the National Enquirer (or equivalent tabloid in Dublin) saying “Scientists Find Alien Civilization” when in fact they only reported locating an unusual array of ancient ruins on a remote island. Whining about media coverage is a straw man. It’s irrelevant to question of whether the scientific literature reveals evidence for epigenetic inheritance, or not. A brief peek into the biology of intergenerational inheritance As I mentioned above, intergenerational inheritance involves a direct stressor to the developing germline. How might that work? To offer an example of just one molecular mechanism, in the early germ cell, exogenous steroid or steroid-like molecules can bind to receptors (or exogenous substances can impede normal binding of endogenous steroids), which can translocate to the nucleus, and act directly on the genome as transcription factors. This may affect chromatin and/or how the DNA gets re-methylated in the germ cells, and after fertilization may cause changes in transcription in adult tissues, via either up- or down-regulation. This is not magical thinking or hocus pocus —as Mitchell seems to perceive it—but boring reproductive molecular biology. From a broader perspective, the development and reprogramming of early germ cells is overwhelmingly complex but appears to involve very precisely choreographed, and heavily evolutionarily conserved, activation and deactivation of many layers of molecular mechanisms, many of which are informed by hormonal or toxicological actions. Interference with this delicate process can persist in the germ cells and reveal themselves as traits or pathologies in the offspring. While Mitchell did not respond to my query to him about vulnerabilities of early germ cells, I imagine he might raise the point that any molecular mischief inflicted on a primordial germ cell is biologically moot due to post-conception epigenetic reprogramming in the form or demethylation and remethylation of the germline. Okay, but as I understand it some regions escape this process, including retrotransposons and some genes associated with metabolic and neurological disorders, and further, other mechanisms could remain in play, for example ncRNAs and histone marks. Long story short, the biology is pretty clear that early exposures can have persistent consequences through the germline. Mitchell cherry picks human studies Concerned about his cherry picking of human studies, I asked Mitchell via Twitter why he ignored human study evidence that diethylstilbestrol given in pregnancy (F0) caused intergenerational (F2) effects. He did not deny this evidence but seemed to backpedal, saying “I was focusing here on the literature claiming effects of social experiences from grandparents to grandchildren.” Putting aside his characterization of starvation as a “social experience,” it’s pretty clear that his blog did not make that distinction—to the contrary, it fist-pounded, “So, there you have it. In my opinion, there is no convincing evidence showing transgenerational epigenetic inheritance in humans.” Nor did he amend or clarify the blog after I brought this issue to his attention. So, no, I don’t believe that is what he meant.[fn] I, perhaps more than anyone, would like to see more studies of environmentally induced inheritance in humans. Why is there not more, though? First, multigenerational human epidemiological cohorts are hard to come by—three generations for intergenerational inquiries are rare, and four generations for transgenerational studies nonexistent. And second, human molecular studies are pretty much impossible due to ethical considerations and lack of access to the tissues of interest. As Mitchell must be aware, we can’t expose pregnant women to toxicants or starvation or whatnot and then extract their fetuses and their germ cells, or carve into their unconsenting babies’ or grandbabies’ brains or body parts. So obviously we must rely on animal models to enrich our understanding of mechanisms. But back to epidemiology, while I can understand some of Mitchell’s concerns about Överkalix, Dutch Famine, and São Gonçalo conclusions—sample size, study design, multiple testing corrections and “noise” in the data are valid points—there are no perfect human retrospective studies. Even with their imperfections, these carefully considered analyses offered early hints about germline-mediated generational effects of developmental-stage stresses, and have an important place in the history of epidemiology. No one ever said they should stand in for all of human epidemiology in this realm of inheritance. But now that it’s 2018, let’s please acknowledge other human studies that have found intergenerational effects of exposures, including the following. Kioumourtzoglou M, et al. Association of Exposure to Diethylstilbestrol During Pregnancy With Multigenerational Neurodevelopmental Deficits. JAMA Pediatr published online May 2018. This study of 47,450 women in the Nurses’ Health Study II found significantly elevated odds for ADHD in the grandchildren of women who took the toxic synthetic hormone drug DES (diethylstilbestrol) during pregnancy. [Yes, some studies I list in this response are new and I know Mitchell is not clairvoyant. Nevertheless they help paint the larger picture.] • Also see JAMA Peds commentary: Nigg, J Toward an Emerging Paradigm for Understanding Attention Deficit Hyperactivity Disorder and Other Neurodevelopmental, Mental, and Behavioral Disorders: Environmental Risks and Epigenetic Associations, JAMA Peds, published online May 2018. (“If disorders like ADHD are epigenetic conditions [that is, dependent on or heavily modulated by discoverable epigenetic changes that are traceable to preventable environmental exposures], it would have powerful implications for where national research dollars should focus to find ways to reduce the incidence of ADHD and other mental disorders.”) Golding J, et al. Grand-maternal smoking in pregnancy and grandchild’s autistic traits and diagnosed autism. Sci Rep 2017;7:46179. This study of the ALSPAC cohort found significantly higher risk of autism and autism-related traits in the grandchildren of women who smoked cigarettes during pregnancy, through the exposed females. Tournaire M, et al. Birth defects in children of men exposed in utero to diethylstilbestrol (DES), Therapie, March 3, 2018. The study suggests an increased incidence of two male genital tract defects in sons of men prenatally exposed to DES. This intergenerational effect had already been observed in animals and in the offspring of women prenatally exposed to DES. Kalfa N, et al. Prevalence of hypospadias in grandsons of women exposed to diethylstilbestrol during pregnancy: a multigenerational national cohort study. Fertil Steril 2011;95(8):2574-2577. This nationwide cohort study in collaboration with a French association of DES-exposed women studied 529 families and showed that a significant proportion of boys born to DES daughters exhibited hypospadias. Brouwers MM, et al. Hypospadias: a transgenerational effect of diethylstilbestrol? Hum Reprod 2006;21(3):666-669. An increased risk of hypospadias was observed when mothers were exposed to DES in utero. However, the excess risk appears to be of much smaller magnitude than in the 2002 study (below). Klip H, et al. Hypospadias in sons of women exposed to diethylstilbestrol in utero: a cohort study. Lancet 2002;359(9312):1102-1107. Found an increased risk of hypospadias in the sons of women exposed to DES in utero, though the absolute risk was small. Titus-Ernstoff L, et al. Menstrual and reproductive characteristics of women whose mothers were exposed in utero to diethylstilbestrol (DES). Int J Epidemiol 2006;35 (4):862-868. Found menstrual irregularity and possible infertility in granddaughters of women who took DES in pregnancy. Titus-Ernstoff L, et al. Birth defects in the sons and daughters of women who were exposed in utero to diethylstilbestrol (DES). Int J Androl 2010;33:377–84. Data suggest a possible association between the mother’s prenatal DES exposure and birth defects in their offspring, particularly in daughters. We cannot, however, rule‐out the possible influence of reporting bias. In particular, the exposed daughters’ elevated risk of cardiac defects may be as a result of the underreporting of these conditions by unexposed mothers. Titus-Ernstoff L, et al. Offspring of women exposed in utero to diethylstilbestrol (DES): a preliminary report of benign and malignant pathology in the third generation. Epidemiology 2008;19:251–7. Based on a small number the risk of ovarian cancer was higher in daughters of women prenatally exposed to DES. Shnorhavorian, M, et al. Differential DNA methylation regions in adult human sperm following adolescent chemotherapy: potential for epigenetic inheritance, PloS One 2017;12(2)journal.pone.0170085. Adolescent chemotherapy exposure promoted epigenetic alterations that persisted approximately ten years after exposure. A signature of statistically significant DMRs was identified in the exposed males, found in CpG desert regions of primarily 1 kilobase size. This study did not investigate phenotypic outcomes in the next generation, but the topic, and suggestion of possible impairment of offspring, is of such tremendous social importance I felt the need to include it here. And here’s an interesting new one, though mechanisms of transgenerational effect are unknown: Patel B, et al. Transgenerational effects of chemotherapy: Both male and female children born to women exposed to chemotherapy have fewer children. Cancer Epidemiology October 2018;56:1-5. The sons and daughters (F1 generation) of chemotherapy-exposed women have fewer (74-77% fewer) live births when compared to both matched, unexposed general population and cousin controls. Of course we would benefit from more evidence of generational effects of exposures in humans. But to say “there is none,” zippo, nada evidence of intergenerational epigenetic inheritance (again, what he really meant by transgenerational), well, that’s just nuts, a reckless, unfair characterization of the science. But worse is his assertion that further research in the area is a waste of time—propped up only by sociological pressures. To the contrary, these studies are delectable hors d’oeuvres to what should be a meatier main course. Now is the time to intensify our inquiries: what are the heritable germ cell effects of pregnancy and neonatal general anesthesia? Of tobacco (aside from the study I cited)? Synthetic steroids aside from DES? Of psychoactive drugs? What about opioids or cannabis? Of heavy doses of industrial chemicals or pesticides? The list of ridiculously important human exposures we should be studying could fill this entire blog. It fairly terrifies me that an influential academic seems to consider these lines of inquiry unimportant. Mitchell ignores abundant evidence of epigenetic inheritance in mammals Okay, this part really got me. Mitchell admits there is very good evidence for epigenetic inheritance in nematodes and plants, and “in specific circumstances involving transposable elements in mice,” linking to a single 2006 study that examined mechanisms in a genetically induced model. Okay, but what about all those other mammal studies in the literature? He contends they “suffer from all the same methodological problems as these human studies, as I have previously discussed here and here" (linking to blog posts that hardly discuss mammal studies). No disrespect, but c’mon, Mitchell’s treatment of this subject is sloppy to the point of awful. Please permit me to share more than 30 of the studies supporting the existence of intergenerational epigenetic, or at least nongenetic (for those observing phenotypes but not mechanisms), inheritance in mammals.
Mitchell has long been vocal with his antagonism toward the concept that exposures can have generational consequences through the germline epigenome. Agents of general anesthesia and analgesics Ju LS, et al. Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats, Brit J Anesth 2018. In a rat model, neonatal exposure to the widely used general anesthetic agent sevoflurane can affect the brains and behavior of the next generation of males through epigenetic modification of Kcc2 expression, while F1 females are at diminished risk. • Also See BJA editorial: Vutskits L, et al. A poisoned chalice: the heritage of parental anaesthesia exposure, Brit. J Anesth, 2018. (“Hence, we are faced with a real possibility that general anaesthetics are not innocuous agents that ‘only put children to sleep’ but rather formidable modulators of chromatin remodeling and function…. The current study extends previous reports of sex differences by showing that anaesthetic exposure itself can alter expression of chloride channels in certain brain regions and that this effect is heritable from exposed male parents to unexposed offspring.”) Chalon J, et al. Exposure to halothane and enflurane affects learning function of murine progeny. Anesth Analg 1981;60:794–7. In a mouse model, learning retardation was seen in offspring of murine parents exposed to GA in utero—in other words, mental impairment in the grandpups of the exposed gestating dams. Tang C-K, et al. Exposure of sires to enflurane affects learning function of murine progeny. Obstet Anesth Dig 1985;5:2, 67. In a mouse model, the general anesthetic agent enflurane administered to male mice was found to adversely affected learning function of their offspring. Rossitto, M, et al, Intergenerational effects on mouse sperm quality after in utero exposure to acetaminophen and ibuprofen. FASEB J. 2018. In a mouse model, demonstrates that pregnancy exposure to these analgesics during the critical period of sex determination affects the germ-line development and leads to adverse reproductive effects in the grandpups. Nicotine and tobacco Zhu J, et al. Transgenerational transmission of hyperactivity in a mouse model of ADHD. J Neurosci 2014;34:2768–73. In a mouse model, grandpups of gestating dams exposed to nicotine exhibited behaviors comparable to ADHD. Rehan VK, et al. Perinatal nicotine-induced transgenerational asthma. Am J Physiol Lung Cell Mol Physiol 2013;305:L501–7. In a rat model, grandpups of gestating dams exposed to tobacco smoke exhibited higher risk for asthma traits. Meier MJ, et al. In utero exposure to benzo[a]pyrene increases mutation burden in the soma and sperm of adult mice. Environ Health Perspect 2017;125:82–8. In a mouse model, higher mutation rates were found in offspring sperm when the pregnant dam was exposed to the tobacco component BaP. Okay slightly off topic, but let’s not forget the importance of de novo mutagenesis in germ cells, which can be precipitated by exposure to mutagenic substances such as BaP. Now back to our program. Synthetic steroids Moisiadis VG, et al. Prenatal Glucocorticoid Exposure Modifies Endocrine Function and Behaviour for 3 Generations Following Maternal and Paternal Transmission. Sci Rep 2017;7:11814. In a guinea pig model, gestational treatment with synthetic glucocorticoids (betamethasone) at a clinically relevant dose resulted in various generational pathology including altered cortisol response to stress, altered expression of genes in the prefrontal cortex and hypothalamic paraventricular nucleus. Transgenerational alterations of programming was seen through F3. Transmission was sex- and generation-dependent, occurring through both parental lines. Iqbal M, et al. Transgenerational effects of prenatal synthetic glucocorticoids on hypothalamic-pituitary-adrenal function. Endocrinology 2012;153, 3295–3307. In a guinea pig model, gestational treatment with synthetic glucocorticoids (betamethasone) modified HPA function and behavior in the F2. Long NM et al. Multigenerational effects of fetal dexamethasone exposure on the hypothalamic-pituitary-adrenal axis of first- and second-generation female offspring. Am J Obstet Gynecol 2013; 208, 217.e1–217.e8. In a sheep model, the synthetic glucocorticoid dexamethasone administed in the clinical range to gestating ewes have multigenerational effects on HPA activity. Drake AJ, et al. Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am J Physiol Regul Integr Comp Physiol 2005;288, R34–R38. In a sheep model, pregnant ewes were exposed to the synthetic glucocorticoid dexamethasone, a variety of pathologies (reduced birth weight, glucose intolerance, and elevated hepatic PEPCK activity) were seen in male grandoffspring. Drake AJ, et al. Multigenerational programming in the glucocorticoid programmed rat is associated with generation-specific and parent of origin effects. Epigenetics 2011;6:1334–43 In a rat model, prenatal glucocorticoid overexposure caused effects on fetal and placental weight in both the F1 and F2 offspring, with marked parent-of-origin effects in F2. Vaughan OR, et al. (Dexamethasone treatment of pregnant F0 mice leads to parent of origin-specific changes in placental function of the F2 generation. Reprod Fertil Dev 2015;27(4):704-11. In a mouse model, effects of F0 gestating dam dexamethasone exposure are transmitted intergenerationally to the F2 placenta via the maternal, but not paternal, line. de Assis S, et al. High-fat or ethinyl-oestradiol intake during pregnancy increases mammary cancer risk in several generations of offspring. Nat Commun 2012;3:1053. In a rat model, fetal exposure to diets high in fat or a large amount of estrogen heightened the risk of breast cancer for three generations of female offspring. Epigenetic changes in the mammary glands of three generations of the rats who had been exposed to increased estrogen were observed. Horan TS, et al. Germline and reproductive tract effects intensify in male mice with successive generations of estrogenic exposure. PLOS Genetics 2017;1006885. In a mouse model, multiple generations of exposure not only exacerbate germ cell exposure effects, but also increase the incidence and severity of reproductive tract abnormalities. Environmental endocrine disruptors/pesticides/plasticizers Crews D, et al. Epigenetic transgenerational inheritance of altered stress responses. Proc Natl Acad Sci USA 2012;109:9143–8. In a rat model, a single exposure to vinclozolin altered the physiology, behavior, metabolic activity, and transcriptome in discrete brain nuclei in descendant males, causing them to respond differently to chronic restraint stress. Wolstenholme JT, et al. Gestational exposure to bisphenol A produces transgenerational changes in behaviors and gene expression. Endocrinology 2012;153:3828–38. In a mouse model, gestational exposure to BPA produces multigenerational alterations in genes and behavior. Drobná Z, et al. Transgenerational effects of bisphenol A on gene expression and DNA methylation of imprinted genes in brain. Endocrinology 2018;159:1132–144. In a mouse model, gestational exposure to BPA produces multigenerational alterations in brain tissues. BPA-caused changes at two imprinted genes in the brain were observed. Gely-Pernot, A, et al. Gestational exposure to chlordecone promotes transgenerational changes in the murine reproductive system of males Sci Rep 2018;8:10274. In a mouse model, pregnant females were exposed to chlordecone, an organochlorine insecticide. Subsequent generations suffered reduction in spermatogonia, meiotic defects in spermatocytes and decrease in spermatozoa number. Changes in the expression of genes associated with chromosome segregation, cell division and DNA repair were observed. Altered epigenetic marks were conserved between F1 and F3 generations. Skinner MK, et al. Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS One 2008;3:e3745. In a rat model, gestating females were exposed to the endocrine disrupting fungide vinclozolin during fetal gonadal sex determination. Alterations to epigenetic reprogramming of the male germ-line and offspring brain transcriptome (sex-specific) were observed, Several brain signaling pathways were influenced including those involved in axon guidance and long-term potentiation. Iqbal K, et al. Deleterious effects of endocrine disruptors are corrected in the mammalian germline by epigenome reprogramming. Genome Biol 2015;16:59. In a mouse model, gestating mice were treated with endocrine-disrupting chemicals vinclozolin, bisphenol A, or di-(2-ethylhexyl)phthalate, resulting in changes in transcription and methylation in the F1 germline. Though intergenerational changes were observed, transgenerational (no direct exposure) were not. Manikkam M, et al. Dioxin (TCDD) Induces Epigenetic Transgenerational Inheritance of Adult Onset Disease and Sperm Epimutations. PLoS One 2012:e46249. In a rat model, gestating females were exposed to dioxin, increasing the incidences of multiple diseases in subsequent generations, including kidney disease in males, pubertal abnormalities in females, ovarian primordial follicle loss and polycystic ovary disease. Analysis of the F3 sperm epigenome identified 50 differentially DNA methylated regions in gene promoters. Valproic acid Choi CS, et al. The transgenerational inheritance of autism-like phenotypes in mice exposed to valproic acid during pregnancy. Sci Rep 2016; 6:36250. In a mouse model, valproic acid (an anti-convulsant drug) induced epigenetic inheritance of autism-like neurobehavioural phenotype in mice through the paternal germline in first and second generation. Cipriani C, et al. High expression of Endogenous Retroviruses from intrauterine life to adulthood in two mouse models of Autism Spectrum Disorders. Sci Rep 2018 8(1):629 In a mouse model, findings of a transgenerational effect of prenatal valproic acid exposure. In the second and third generation, more marked transcriptional effects were seen in offspring from females, compared to paternal lineages. McBirney M, et al. Atrazine induced epigenetic transgenerational inheritance of disease, lean phenotype and sperm epimutation pathology biomarkers. Plos ONE 2017;12(9): e0184306. https://doi.org/10.1371/journal.pone.0184306 In a rat model, gestating females were exposed to atrazine. The F2 generation (grand-offspring) was found to have increased frequency of testis disease and mammary tumors in males and females, early onset puberty in males, and decreased body weight in females compared to controls. Chemotherapeutic agents Glen, CD, et al. Exposure to anticancer drugs can result in transgenerational genomic instability in mice. Proc Natl Acad Sci 2012;109(8):2984–2988. In a mouse model, paternal exposure to a commonly used chemotherapeutic agents resulted in increased mutation rates and transgenerational instability. (De novo mutagenesis, not epigenetic per se, was investigated.) Chan, D et al. Epigenetic alterations in sperm DNA associated with testicular cancer treatment. Toxicol Sci 2012;125(2):532–543. In a rat model, treatment with chemotherapeutic agent resulted in DNA methylation alterations in sperm. This study did not investigate resulting phenotype in offspring borne of the epigenetically altered sperm. Diet Dunn, GA et al. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 2011;152, 2228–2236. In a mouse model, a high-fat diet in gestation resulted in larger F3 female offspring. A potential dynamic pattern of paternally expressed genes from the paternal lineage was seen at an imprinted locus, thereby providing sex specificity to both the transmission and inheritance of traits related to disease predisposition. Odor fear conditioning Dias, BG et al. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat Neurosci 2014;17, 89–96. In a mouse model, pre-conception adult mice were subjected to odor fear conditioning, with behavioral, neuroanatomical, and epigenomic consequences in unexposed pups and grandpups. Immune activation Weber-Stadlbauer, U. et al. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol Psychiatry 2017; 22:102-112. In a mouse model, prenatal immune activation by the viral mimetic poly(I:C) resulted in alterations in brain and behavioral functions in multiple generations. Reduced sociability and increased cued fear expression in first- and second-generation offspring were observed, in addition to other sex-specific effects. Genome-wide transcriptional changes were also seen. Chronic stress/traumatic experience Rodgers, AB, et al. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci 2013;33:9003–9012. In a mouse model, males were exposed to six weeks of chronic stress before breeding. Offspring displayed significantly reduced HPA stress axis responsivity. Changes in transcription were seen in the brain, suggestive of epigenetic reprogramming and consistent with altered offspring stress responsivity, including increased expression of glucocorticoid-responsive genes in the PVN. In examining potential epigenetic mechanisms of germ cell transmission, robust changes in sperm microRNA were found. Rodgers, AB, et al. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci USA 2015;112,13699–13704. In a mouse model, after males were exposed to chronic stress, sperm miRNAs were found postfertilization to alter offspring stress responsivity. Also zygote microinjection of the miRs, demonstrated a recapitulation of the offspring stress dysregulation phenotype. Gapp, K. et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci 2014;17, 667–669. In a mouse model, traumatic stress in early life altered mouse microRNA expression, and behavioral and metabolic responses in the progeny. Injection of sperm RNAs from traumatized males into fertilized wild-type oocytes reproduced the behavioral and metabolic alterations in the resulting offspring. Franklin, TB, et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry 2010; 68, 408–15. In a mouse model, mice were exposed to chronic and unpredictable maternal separation from postnatal day 1 to 14. Alterations in the profile of DNA methylation in the promoter of several candidate genes in the germline of the separated males were observed. Comparable changes in DNA methylation are also present in the brain of the offspring and are associated with altered gene expression. Weiss, IC, et al. Inheritable effect of unpredictable maternal separation on behavioral responses in mice. Front Behav Neurosci 2011; 5,3. In a mouse model, unpredictable maternal separation from birth to postnatal day 14 in C57Bl/6J mice has mild behavioral effects in the animals when adult, but that its combination with maternal stress exacerbates this effect. Further, the behavioral deficits are transmitted to the following generation through females, an effect that is independent of maternal care and is not affected by cross-fostering. The combined manipulation does not alter basic components of the hypothalamic–pituitary–adrenal axis but decreases the expression of the corticotropin releasing factor receptor 2 (CRFR2) in several nuclei of the amygdala and the hypothalamus in the brain of maternal-separated females. Gapp, K. et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 2014. doi:10.1038/nn.3695 In a mouse model, injection of sperm RNAs from traumatized males into fertilized wild-type oocytes reproduced the behavioral and metabolic alterations in the resulting offspring. Bohacek, J. et al. Pathological brain plasticity and cognition in the offspring of males subjected to postnatal traumatic stress. Mol. Psychiatry 2014; doi:10.1038/mp.2014.80 In a mouse model, males subjected to postnatal traumatic stress have offspring with defects associated with impaired long-term memory. The expression in offspring of brain-specific signaling component in the hippocampus is reduced in the offspring, and DNA methylation at its promoter is altered both in the hippocampus of the offspring and the sperm of fathers. Razoux, F. et al. Transgenerational disruption of functional 5-HT1AR-induced connectivity in the adult mouse brain by traumatic stress in early life. Mol. Psychiatry 2016. doi:10.1038/mp.2016.146 In a mouse model, traumatic stress in postnatal life alters 5-HT1A receptor-evoked local and global functions in progeny of the exposed animals. Disrupted functional connectivity is consistent across generations and match limbic circuits implicated in mood disorders. Mechanism papers It’s also worth including a few other papers exploring mechanisms of nongenetic inheritance in mammals. Zhang Y, et al. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat Cell Biol 2018; 20(5):535-540. Benito E, et al. RNA-Dependent Intergenerational Inheritance of Enhanced Synaptic Plasticity after Environmental Enrichment. Cell Rep 2018; 23(2):546-554. Dickson DA, et al. Reduced levels of miRNAs 449 and 34 in sperm of mice and men exposed to early life stress. Translational Psychiatry May, 2018. Sharma U, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 31 January 2015. Huypens P, et al. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat Genetics 2016;48:497-499. Grandjean V, et al. RNA-mediated paternal heredity of diet-induced obesity and metabolic disorders. Sci Rep 2015;5:18193. So. is there really such a dearth of findings in mammal studies in epigenetic inheritance? Really? Really?! Can any reasonable person interpret the studies and conclude “no convincing evidence of intergenerational epigenetic inheritance in mammals”? How is that even possible? Wouldn’t a more reasonable response to the literature be, “Golly, such a plethora of evidence of exposure-induced intergenerational epigenetic inheritance in mammals, this sure looks like a phenomenon we should take rather seriously”? Or, to borrow a line from the 2006 paper cited by Mitchell, “Non-Mendelian inheritance, such as this, is of particular interest if it exists in humans, since it would have a profound effect on the inheritance of phenotypic traits.” [By the way, for the uber-curious, I list several reviews of these topics in Appendix 2.] Conclusion Sloppy overstatement and dogmatism from the Ivory Tower, such as Mitchell’s blogpost, can breed complacency precisely at a time when we should be deeply alarmed about the intergenerational effects of past and current exposures. It should be clear to all of us by now that molecular insults to the germline can influence disease, behavior or physiology of offspring, perhaps in ways that are staggeringly important for public health. While healthy skepticism is always welcome, research does not progress by allowing outspoken academicians to distort the state of the science, unchallenged.
Appendix 1: Overview of commonly referenced critical windows for nongenetic inheritance, by sex A. Exposures to females 1. Exposure to an adult gestating female. This exposure involves a direct exposure to a gestating F0 female, the F1 embryo/fetus and the F2 embryonic/fetal germ cells residing within. (a) The directly exposed female is the F0, implying possible impacts to her somatic cells. (b) The directly exposed male or female embryo/fetus is the F1 child soma. This is usually referred to as a “prenatal” or “fetal” exposure. Because it involves only somatic effects it is not considered part of epigenetic inheritance. (c) The male or female F1’s directly exposed embryonic/fetal germ cells (F2) later yields male or female grandchildren, or F2 genotype/epigenotype and F2 phenotype. This is usually referred to as “intergenerational” inheritance. Most of the studies cited by Mitchell involve intergenerational, and not transgenerational, inheritance. Because embryonic/fetal germ cells are highly dynamic featuring multiple levels of reprogramming (more on this later) this stage is considered an important critical window for exposure effects. (d) The F3 are the great-grandchildren of the gestating female. Effects of any F0 gestational exposure are considered “transgenerational” involving no direct exposure either to the adult F2’s germline or F3 soma. None of the studies cited by Mitchell examine this transgenerational effect, though some use the word “transgenerational” to generally mean anything past an F0 impact. 2. Exposure to a neonate female. Because the delicate process of genomic imprinting continues in the female ovary well after birth and through the first year, the infancy of the female may also present a critical window for exposures, though the molecular impacts are likely to be different than those seen in the primordial germ cell. An impact on the next generation borne of those infancy-exposed eggs would be considered intergenerational. 3. Exposure to a slow-growth period female. The period before puberty. Next generation effects would be considered “intergenerational.” 4. Exposure to an adult pre-conception, non-gestating female. Some studies consider stressors to an adult female in the period well before or shortly before conception. I don’t see a lot of evidence this window is a critical period of vulnerability for the egg epigenome, but I could be wrong. That said, a pre-conception exposure of interest need not occur just before conception to be biologically relevant. A drug or chemical can be slow to metabolize, or may lodge in the fatty tissues even for decades (think of DDT or dioxin, for example). So long-gone exposures may still directly impact the ovulated egg (intergenerational), the conceptus (somatic), or the developing embryo (somatic). Real-life biology is complicated, alas. B. Exposures to males Because males don’t gestate babies, and generate sperm instead of eggs, male-line epigenetic inheritance is a different ballgame. 1. Exposure to germ cells of a F1 embryonic/fetal male gestating in F0 female. This was discussed in A(1)(c), above. Offspring of F1 germ cells effects would be considered “intergenerational” (grandchild of the exposed gestating female). The following generation, great-grandchild (F3) of gestating female effects would be “transgenerational,” involving no direct exposure to those germ cells or soma. 2. Exposure to a neonate male. Similar to A(2), above, except that the male germ cells are apparently done with the imprinting process before birth. Next generation effects would be considered “intergenerational.” The following generation effects would be “transgenerational.” 3. Exposure to a slow-growth period male. The period before puberty. Next generation effects would be considered “intergenerational.” 4. Exposure to an adult pre-conception male. The final phase of spermatogenesis, the approximately 72 days when primary spermatocyte matures into sperm. Next generation effects would be considered “intergenerational.” The same complex considerations in A(4) above apply here. Appendix 2: Some reviews of interest Bale, TL. Epigenetic and transgenerational reprogramming of brain development. Nat Rev Neurosci 2015;16:332–344. Pembrey, M, et al. Human transgenerational responses to early-life experience: potential impact on development, health and biomedical research. J Med Genet 2014;51:563–572. Alonso-Magdalena, P, et al. Bisphenol-A and metabolic diseases: epigenetic, developmental and transgenerational basis. Environ Epigenetics 2016:2. Xin, F, et al. Multigenerational and transgenerational effects of endocrine disrupting chemicals: A role for altered epigenetic regulation? Semin Cell Dev Biol. 2015;43:66–75. Ozgyin, L, et al. Nuclear receptors in transgenerational epigenetic inheritance. Prog Biophys Mol Biol 2015;118:34–43. Skinner, M. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol. Cell. Endocrinology 2014;398:4–12. Dunn, GA ,et al. Sex-specificity in transgenerational epigenetic programming. Horm Behav 2011;59:290–295. Gapp K, et al. . Epigenetic germline inheritance in mammals: looking to the past to understand the future. Genes, Brain and Beh 2018;17:3,e12407. Soubry A, et al. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. Bioessays 2014;36:359–71. Soubry, A. POHaD: why we should study future fathers. Environ Epigenetics 2018; 4:2. Bohacek, J, et al. Epigenetic Inheritance of Disease and Disease Risk. Neuropsychopharmacology 2013;38:220–236. Bohacek J, et al, Transgenerational Epigenetic Effects on Brain Functions, Biol Psych 2013;73(4) 313-320. Barlow DP, et al. Genomic imprinting in mammals. CSH Perspect Biol 2014;6(2). And for those seeking an even longer read, a newly published book: Bonduriansky and Day, Extended Heredity, Princeton 2018. Jill Escher is a research philanthropist who funds pilot studies investigating exposure-induced nongenetic inheritance (Escher Fund for Autism). She is also president of Autism Society San Francisco Bay Area, a housing provider to adults with developmental disabilities, a former lawyer, and the mother of two children with idiopathic nonverbal autism. Learn more about her research philanthropy and science advocacy at GermlineExposures.org.
1 Comment
Escher Fund for Autism 1590 Calaveras Avenue San Jose, CA 95126 (408) 314-1655 jill.escher@gmail.com germlineexposures.org Division of Dockets Management Food and Drug Administration Department of Health and Human Services 5630 Fishers Lane, rm. 1061 Rockville, MD 20852 February 5, 2018 Re: Docket Number FDA-2015-P-0876—Re Citizen Petition to Withdraw Approval for 17α-Hydroxyprogesterone Caproate (“17-OHPC,” Including Brand “Makena”) as a Drug Used in Pregnancy, Pending Fetal Germline Impact Assessment To the Commissioner of the Food and Drug Administration: The FDA received the undersigned’s above-referenced petition on March 19, 2015. Having received no response after nearly three years, the petitioner again asks that the FDA act expeditiously to grant the petition. (The petition can be found online here.) The petitioner’s February 2017 letter entreating to the FDA to take some action to consider the petition emphasized the unequivocal drug vulnerabilities of fetal germline to intentional, high-dose persistent EDC exposures such as 17-OHPC. However, other urgent and perhaps more obvious reasons to withdraw approval for 17-OHPC have also surfaced, including the following: (1) 17-OHPC is ineffective at reducing preterm birth. The evidence is now clear that administration of 17-OHPC is ineffective at reducing risk for recurrent preterm birth. In keeping with previous findings from other studies, a 2017 prospective cohort study demonstrated this synthetic hormone did not reduce the rate of recurrent preterm birth. (Nelson DB, McIntire DD, McDonald J, et al. 17-alpha Hydroxyprogesterone caproate did not reduce the rate of recurrent preterm birth in a prospective cohort study. American Journal of Obstetrics and Gynecology, 2017;216(6):600.e1–600.e9 [also discussing how earlier research failed to document a benefit of this drug]). (2) 17-OHPC increases risk of maternal diabetes in the exposed mother. The above-mentioned study also found the drug increased the risk for the mother developing gestational diabetes. Id. This represents a significant adverse outcome that is not included in warnings given the patient before administration of this hormone-disrupting chemical. (3) 17-OHPC causes long-term adverse neurodevelopmental effects in offspring. It is well documented that perturbations of sex steroids can adversely affect neurodevelopment in affected fetuses. (Gore AC, Martien KM, Gagnidze K, Pfaff, P. Implications of Prenatal Steroid Perturbations for Neurodevelopment, Behavior, and Autism. Endocr Rev. 2014;35(6):961–991.) Many regions of the developing brain are sensitive to progestins, including neural circuits important for complex cognitive behaviors later in life. (Willing J, Wagner CK. Exposure to the Synthetic Progestin, 17α-Hydroxyprogesterone Caproate During Development Impairs Cognitive Flexibility in Adulthood. Endocrinology 2016;157(1)1:77–82.) A recent study (Id.) documented adverse long-term consequences of 17-OHPC exposure during development on cognitive behavior of offspring. Administration of the drug to gestating rats led to dopaminergic innervation of specific lamina of prelimbic mPFC in juveniles, consistent with the possibility of impaired synaptic pruning. Additionally, 17-OHPC exposure impaired cognitive flexibility in adulthood—neurocognitive effects reminiscent of those often associated with developmental disorders such as attention-deficit hyperactivity disorder and autism. (Id.) The-treated rats were slower to make a cognitive switch to a new rule and continued to perseverate on the old rule longer than controls. (Id.) Abnormal levels of progesterone receptor activity and/or progesterone receptor activity at improper times during this critical period of connectivity may significantly alter the development of important behavioral neural circuits. (Id.) (4) A safer alternative to 17-OHPC is readily available. Daily vaginal progesterone (either suppository or gel) started at about 16 weeks’ gestation has been found to be a preferable alternative to weekly 17-OHPC injection for prevention of spontaneous preterm birth in women with singleton gestations and prior spontaneous preterm birth. (Saccone G, Khalifeh A, Elimian A, Bahramis E, et al. Vaginal progesterone vs intramuscular 17α-hydroxy- progesterone caproate for prevention of recurrent spontaneous preterm birth in singleton gestations: systematic review and meta-analysis of randomized controlled trials. Ultrasound Obstet Gynecol, 2017;49: 315–321.) Compared with intramuscular 17-OHPC, vaginal progesterone was associated with at least seven benefits: (1) reduced risk of recurrent SPTB; (2) fewer adverse maternal side effects; (3) fewer NICU admissions; (4) lower cost; (5) better maternal compliance; (6) women’s preferred choice; and (7) greater satisfaction. (Id.) In closing, the FDA was created by Congress to safeguard the public from pharmaceutical products that confer more risk than benefit, yet today, in spite of ever-mounting evidence that 17-OHPC is an ineffective and dangerous gestational toxicant, the agency does nothing but sit on its hands in a seemingly permanent state of paralysis. The diethylstilbestrol (DES) catastrophe has already alerted the FDA to the reality that minute perturbations inflicted by synthetic steroid hormones on developing fetuses can cause outsize, horrific long-term effects. The time to withdraw the FDA’s misguided approval for 17-OHPC is now. The health and welfare of three generations is at stake: the exposed mother, the exposed offspring, and per my original petition, the exposed fetal germ cells. Thank you again for your consideration of this petition. Though three years have passed I still eagerly await your response. Very truly yours, Jill Gilbert Escher Cc: Robert Heflich, PhD, Director, Division of Genetic and Molecular ToxicologyUS FDA Rosalie Elespuru, PhD, Research Biologist, CDRH/OSEL/DBCMSUS, US FDA Manju G Manjanatha, PhD, Division of Genetic & Molecular Toxicology, US FDA Thomas Gellhaus, MD, president, American College of Obstetricians and Gynecologists Alfred Abuhamad, MD, president, Society for Maternal Fetal Medicine  UPDATE: Here is the FDA's February 2018 ruling on the March 2015 petition (pdf). Commentary on this is forthcoming.  Were pioneering scientists who warned of a possible "genetic emergency" prescient geniuses or Chicken Littles? By Jill Escher Fifty years ago, around the time of the founding of the Environmental Mutagen Society (EMS), preeminent biologists, geneticists and toxicologists warned of a potential “genetic emergency.” This alarm referred to the possibility that exposures to novel chemicals and drugs flooding the market in the ebullient post-war era could be wreaking silent havoc in human germline, thereby weakening the developmental integrity of future generations. For example, in 1969 mutagenesis pioneer Sam Epstein warned, “At this moment we may be be in the midst of a potentially serious accidental experiment on the effects of chemical mutagens in man, the full impact of which may not be known for generations to come.” Nobel laureate Joshua Lederberg, knowing of germ cell vulnerability from animal models, in 1955 urged that “more extensive studies are needed to establish … whether germ cells of man are physiologically insulated against … chemical insults from the environment.” It was geneticist James Crow who in 1968 flat-out warned of chemicals causing a future “genetic emergency.” In “Chemical Risk to Future Generations,” he explained the possibility that “some compound presumed to be innocuous is in fact highly mutagenic and that large numbers are exposed before the danger is realized.” 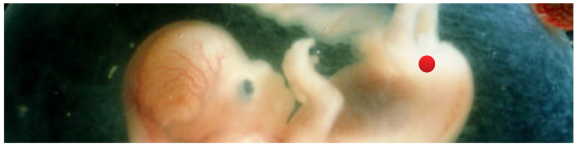 Were these men (and there were women, too, notably the mutagenesis researcher Charlotte Auerbach) and their colleagues prescient geniuses or Chicken Littles? (Fn 1) Have their fears about human germ cell damage come to pass? Or are our gametes so robustly resilient that the onslaught of post-war exposures bounced off them like so many ping pong balls? I think this is one of the greatest scientific and public health questions of our time. Clearly we aren’t seeing visible bad-movie-style mutants roaming our streets and crowding our schools. Cases of three-armed boys and seven-foot girls do not seem be on the rise. But what about more subtle, or at least less visible, phenotypes possibly resulting from mutated or otherwise damaged germ cells? Are we seeing an increase there? I would argue this is quite possibly the case. Over the past several decades we have seen staggering increases in somewhat invisible but often extravagantly disabling disorders of development, including autism, ADHD and learning disabilities. The prevalence of the paradigmatic of these pathologies, autism, has surged more than 20-fold in California since births in the 1980s, for example, and that’s counting only the more severe cases—now fueling a stupendous public health and social services crisis. It is frustrating, then, that autism causation research has been stuck in the dated paradigm of “genes” or “environment.” In the world of autism at least, the teachings of environmental mutagenesis (also referred to as genetic toxicology) seem to have fallen on deaf ears. The bulk of research funds have been spent on rather reductionist gene-hunting, with a lesser sum on potential impacts on fetal soma of environmental exposures, such as pregnancy drugs, adverse nutritional states, or pollutants. The idea that exogenous weird stuff could have damaged germline DNA or any of its many regulatory layers is barely on the radar. Certainly, the idea was barely mentioned during the 2017 International Meeting for Autism Research. At the same time, we are finding a plethora of heterogenous de novo mutations related to the condition. While researchers are quick to attribute these glitches to nothing more than random chance, I say not so fast. Autism is strongly heritable—among siblings. Yet there is essentially no evidence aside from some speculative statistical analyses that autism is handed down through the generations (such as this recent analysis by Wigler and colleagues). And these analytical approaches omit any potential role for exogenous perturbations of gene function; it’s as if the possibility never entered the minds of the researchers. But from a real-biology perspective there would be increased risk in siblings if at least one parent had suffered an adverse germline exposure, affecting genome, epigenome or cytoplasmic element such as mitochondria (ooocyte only), upping the risk for de novo heritable disease in multiple offspring. Strong heritability rates such as those seen in autism can not logically follow from random mutation, but rather from disruptions blanketing parental germ cells (and particularly, I would argue, early germ cells). Nevertheless, the possibility is seldom if ever discussed among geneticists. I would like to argue those doomsayers of the 1950s and 60s, those scientists who warned of a genetic emergency, may well have been prescient geniuses, and not Chicken Littles. Perhaps to a considerable extent today’s mysterious epidemics of neurodevelopmental pathology like autism have arisen as a consequence of silent damage in the human germline, inflicted by chemical forces thought at the time to be fairly innocuous (like, perhaps, my own fetal 1965 synthetic hormone drug exposures). Not ancestral “genes” alone. And not “environment” alone acting on fetal soma. While those single-dimension factors are of course important, why are we neglecting the founding EMS idea, now re-animated through the emerging field of germline epigenetics, that environment can alter our germline? This, I believe, is the forgotten realm of public health sciences. In our over-simplified approach, an entire dimension of biological risk has been neglected. This, I believe, is the forgotten realm of public health sciences. In our over-simplified approach, an entire dimension of biological risk has been neglected. Now, having been in the business for several years of identifying and funding opportunities to examine this outsider hypothesis, I am painfully aware of the difficulties inherent in generational, or germline, studies. In human cohorts, records of 1950s-1970s grandmaternal (F0) gestational exposures (such as to tobacco smoking or pharmaceutical drugs such as the soup of fake hormones to which I was prenatally exposed) are hard to find, and when they do exist, the records of grandchild (F2, sprung from exposed F1 fetal germline) may not be available. And then there are the confounds and complexities of any multigenerational study. Further, many tissues is of interest, including a mother’s eggs, are inaccessible. And a father’s sperm? There’s only been one study in that regard, and while it found differences in the autism father sperm methylome, it did not inquire into past exposures. In the animal model realm, developmental pathologies borne of early germline exposures are often readily seen, but people tend to question the applicability of those studies to humans. As autism epidemiologist Craig Newschaffer remarked to me, autism causation research "has a low-hanging fruit problem." And he's right. Why dig deep to look for old, forgotten exposures and 20 to 50 years of data, when you can simply write the umpteenth paper on paternal age (now that's low-hanging data)? As one epidemiologist remarked to me, autism causation research "has a low-hanging fruit problem." In spite of the logistical and analytical hurdles, germline exposure studies are certainly feasible, and in my view of course they must be done. The first study to be published on this issue relied on the rare trove of generational data of the ALSPAC cohort at University of Bristol in the UK. The study, funded by Escher Fund, found a link between grandmaternal pregnancy smoking and autism traits and diagnosed autism in grandchildren, through the exposed female parents (ie, early oocytes). While some journalists (for example here), found this link “baffling,” anyone cognizant of genetic toxicology would have little problem connecting plausible toxicological and biological dots. It's as if environmental mutagenesis has lapsed from the public consciousness. Fifty years ago mutagenesis was a young field. But today we have the benefit of many scientific insights that the pioneers could not have imagined. We now know, for example, that tobacco smoke is mutagenic. (Talk about something once presumed innocuous, I can only imagine the smoke-filled rooms of the early EMS meetings….). We know that synthetic hormone mimics, also known as endocrine disruptors, can exert adverse generational effects. We know of the importance of timing, and that primordial germ cells have heightened molecular vulnerabilities. And we increasingly know of multiple cellular mechanisms, well beyond classic exome mutations, that can exert heritable effects. This includes regulatory DNA, methylation and genomic imprinting, other epigenomic artifacts such as histone modifications and ncRNAs, and cytoplasmic structures such as mitochondria, and even possible receptor effects not touching DNA directly at all. DNA is but one piece of molecular heritability. "DNA is but one piece of molecular heritability."  Given the tremendous advances in understanding early germ cell vulnerabilities (hey, and even late phases, too, as seen in this recent review of tobacco mutagenicity and adult phase sperm), I find it regrettable that public health research, and regulatory approaches as well, have overlooked this precious and powerful, if invisible, phase of the human lifecycle. There are few studies examining impacts of drugs or smoking on early germ cells, and regulatory approaches overlook them altogether. (Fn 2.) I do wish we will someday heed the words of those EMS pioneers, and geneticist James Neel, who in 1969 summed up what is at stake:
And what are we doing to protect, or even consider past exposures to, mankind's most vital asset? Well, today, I daresay, almost nothing. This is truly the forgotten realm of public health research. Notes: (1) EMS founders included Drs. Alexander Hollaender, Joshua Lederberg, James Crow, Ernst Freese, James Neel, William Russell, Heinrich Malling, Frederick J. de Serres, Matthew Meselson, among others. The EMS is now known by the name Environmental Mutagenesis and Genomics Society. (2) FDA risk assessment of pregnancy drugs, for example, omits any mention of potential impacts on fetal germline. This has been the subject of two Citizen Petitions filed by the Escher Fund for Autism. Jill Escher is the founder of the Escher Fund for Autism. In 1965 she was exposed in utero to continuous and heavy doses of synthetic steroid hormone drugs, a fact she discovered only in 2011. As the mother of two children with severe idiopathic autism, she has found many other affected families that share her exposure story, and has funded pilot research projects to examine possible associations between gestational toxicants, germ cell exposure, and adverse neurodevelopmental outcomes.  A few of the headlines after the study was published in Scientific Reports. While some headlines seemed a bit un-sober, it was gratifying to see the concept of fetal germ cell exposures hit the popular press. 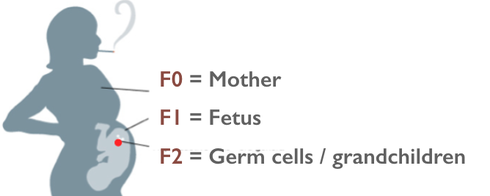 A pregnancy exposure affects two developing generations simultaneously: the fetus and the future grandchildren, via the exposed germ cells. A new UK study funded by the Escher Fund for Autism finds a grandmother's pregnancy smoking elevates risk of autism in grandchildren via the maternal line. By Jill Escher I'm excited to announce the results of a new study, "Grand-maternal smoking in pregnancy and grandchild’s autistic traits and diagnosed autism," published today in Scientific Reports. The study finds an increased risk of autism and autism-related traits in grandchildren, now about 26 years old, of women who smoked during pregnancy. The group studied is the ALSPAC Children of the 90s cohort, led by pioneering epidemiologists Jean Golding and Marcus Pembrey of the University of Bristol. Although the number of grandchildren with autism was small, around 170, the researchers found a notable increased risk of autism and autism-related traits when the grandmother had smoked cigarettes when pregnant with the mother (and, therefore, the mother's early eggs). See the University of Bristol's media release here for further details. Findings include that if a girl's maternal grandmother smoked during pregnancy, the girl is 67% more likely to display certain traits linked to autism, such as poor social communication skills and repetitive behaviors. Also, if the maternal grandmother smoked, this increased by 53% the risk of her grandchildren having a diagnosed autism spectrum disorder (ASD). The Escher Fund for Autism funded the study pursuant to its 2015 grant program, "20th Century Maternal Smoking: Induced Fetal Germline Perturbations in the Etiology of Autism and Neurodevelopmental Disorders.” A growing number of researchers are recognizing the importance of germline exposures, mutagenesis and epimutagenesis when assessing origins of neurodevelopmental pathology. "One of the important insights from this study is the implicit suggestion that modification of the genetic material of germ cells, in this case DNA of eggs, in grandmothers who smoked cigarettes during pregnancy can cause autistic phenotypes in the grandchildren," says Pradeep Bhide, PhD, Professor of Developmental Neuroscience at Florida State University. "Experimental animal models have suggested that epigenetic modification of germ cell DNA due to exposure to nicotine or other chemicals correlates with behavioral impairment in the descendants. Now this multi-generational human study underscores this point quite emphatically – in people." Personally, I see this study as something of a small ship that has struck a vast, unexplored continent. We are accustomed to thinking of autism risk in dualistic terms of genes or prenatal environment. In many cases, however, the answer may hinge instead on a third route: long-ago (and, alas, long forgotten) exposures disturbing elements within our germline genome or other germ cell components. Some of the many questions the study raises include: Do some genetic findings in autism have environmental roots? Perturbations to the germline may cause mutagenesis, epigenetic glitches or cytoplasmic error in the germline. Autism research tends to presume the great variety of genomic glitches seen in autism cases are "random," and that the heritability of autism somehow has natural roots, but has not yet investigated potential exogenous sources of those germline-borne errors. As Dr. Bhide says, "I hope that this new evidence will provide impetus to research on how exposure of the developing fetus to environmental influences, whether cigarettes, hormones or other chemical substances, that may have occurred in generations past can contribute to developmental disorders such as autism in the present generation, even in the absence of a genetic predisposition” Population and demographic effects? While enhanced risk may be relatively small on an individual level, the risk could be significant on a population level, given the large numbers of women who smoked during the latter half of the 20th century, particularly in western countries. and could help explain the timing of the autism increase, as well as the uneven socio-demographic patterns. Does pregnancy smoking also raise risk of autism in grandoffspring through the male line? This pilot study did not detect an association between grandmaternal tobacco use and autism risk through the fathers who had been exposed in utero but that may be due to the limited size of the study sample and not to any true lack of association. Many human and animal studies have demonstrated various deleterious impacts of tobacco on sperm, so this question remains worth pursuing in larger cohorts. A comprehensive literature review by Health Canada researchers is currently in press, examining the effects of smoking on sperm count and quality, chromosome and DNA damage, mutations, and potential impacts. According to the paper, the weight of evidence indicates that smoking impacts all of these parameters. They assessed the potential population level impacts of a modest increase in heritable mutations (25%). "Using 700 genes that are linked to intellectual disease, our model suggests that millions of individuals could be impacted by ID per generation globally as a result of Paternal Smoking," said Carole Yauk, PhD, Research Scientist in Genomics at Health Canada. This estimate is based on 2.4 million non-silent mutations in ID genes worldwide per generation: with 1.9 million being loss-of-function mutations. "This does not include the thousands of extra cases of aneuploid offspring that are likely because of paternal smoking, or deleterious mutations in genes associated with other disorders," she said. The researchers found that costs of this are on the order of $500 billion per generation (conservatively) for ID alone. (Aneuploidy refers to an abnormal number of chromosomes in a cell.) What are the relevant mechanisms of germline error and heritability? During fetal gametogenesis the molecular instruction book for the development of the following generation is in large part written. Research has shown early germline to be vulnerable to toxicants and environmental stressors during this period, affecting DNA, regulatory elements, epigenome, and cytoplasmic elements, including mitochondria. This study did not investigate mechanisms, but follow-up studies should address this question. Implications for regulatory review? Toxic, hormone-signal disrupting, and otherwise geno-affective pregnancy exposures increased dramatically in the post-war decades. These included the surge of maternal smoking and a great variety of synthetic pregnancy drugs. Every pregnancy exposure hits three generations at once: the mother (F0), the fetus (F1), and the future grandchildren (F2) via the exposed fetal germ cells. In spite of this plain biological reality, agencies such as the FDA have never assessed pregnancy drugs or tobacco for effects on the fetal germline or development of the F2 generation. (The Escher Fund for Autism has petitioned the FDA to change this policy of non-consideration of germline, for example here, but so far without effect.) Enduring effects on the subsequent generation? Animal models suggest multigenerational effects of gestational exposures to toxicants and endocrine disrupting chemicals. If tobacco smoke has destabilizing effects on human germline, do these pathological effects possibly endure for generations? This question could be of tremendous significance for autism families and others affected by the deleterious genomic effects of tobacco smoke. Can germline exposures like tobacco help explain the "broader autism phenotype"? Research shows that autism risk is elevated among siblings, but so is the "broader autism phenotype," which refers to related or subclinical traits associated with autism. The results of the ALSPAC study suggest tobacco toxicity could raise risk for both the pathology of autism and also the variety of traits associated with the disorder. What is the significance of doses, timing of smoking? Are only the grandoffspring of heavy (pack a day or more) smokers affected? According to some histories, "chain smoking" by women picked up only after World War II, owing the social factors, marketing and the costs of cigarettes. In addition, does timing of exposure matter? The early germ cell is molecularly vulnerable owing to the dynamic remodeling of the germline, including demethylation and the laying of imprints. The extent of genomic damage may hinge not only on dose but also timing of the exposure. What about other sources of fetal tobacco exposure? Might pre-pregnancy smoking also be significant for the fetus and germ cells, because some components of tobacco remain in maternal tissues long after the cessation of smoking? And what about second-hand smoke, which is also toxic? Further, of the 100s of toxic components of cigarette smoke, which one might be of most concern? For example, Benzo(a)pyrene? Nicotine? Why neurodevelopment? Of all possible phenotypic consequences of germline exposure, why does neurodevelopment seem to be particularly vulnerable? Research suggests a variety of possibilities: the hyper-mutability of long genes associated with neurodevelopment; impairment of genomic imprinting (lasting methylation of a subset of genes, many of which are associated with brain development and function); the role of ncRNAs; the sheer number of genes associated with brain function; and that genes that "escape" or resist epigenetic reprogramming relate to neurological function, among others. But this question was not addressed of course in this epidemiological study. Sex differences? Further examination is needed of differential sex effects on exposed F1 germ cell genome and epigenomic factors. Oogenesis and spermatogenesis feature many key differences in molecular and cellular processes and timing (girls are born with their eggs largely developed, boys are born with spermatogonial stem cells, which mature at puberty and regenerate sperm over many decades). Importantly, egg and sperm feature stark differences in cell physiology and components, which could influence effects of toxicant exposure. Furthermore, sex differences in F2 offspring autism rates and phenotypes warrants study. 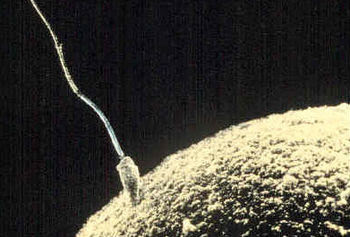 When it comes to heritable factors, egg and sperm are not equivalent. The egg contains vastly more material —and molecular information— carried into the next generation. Exposures during early oogenesis can affect that material. Sperm are also vulnerable through gametogenesis, but via somewhat different mechanisms.
Other pregnancy exposures? There are many additional fetal germline exposures of interest, including pregnancy drugs (for example, I, the mother of two children with severe idiopathic autism, was prenatally exposed to heavy and sustained combinations of synthetic steroid hormone drugs once popular in certain private clinics for ostensible prevention of miscarriage). While I have seed-funded some studies, larger wallets than mine are needed. Many thanks to the ALSPAC research team, especially Drs. Golding and Pembrey, for their assiduous and groundbreaking work, and undertaking this study to help to shed important new light on the forgotten histories that may be influencing autism risk through germ cells. This team has long been at the forefront of appreciating and demonstrating generational impacts. I also wish to thank all the wonderful Children of the 90s families — especially the autism families — for the immense gifts they continue to bestow to the rest of us. Related links: Germline Exposures (Escher Fund for Autism website) To Understand Autism, Talk to the Grandmothers (2017 commentary with background behind hypothesis) Autism's missing link? Study family history alongside genetics (2016 Autism Speaks blog) Out of the Past: Old Exposures, Heritable Effects, and Emerging Concepts for Autism Research (2016 presentation about the hypothesis) Germline Disruption Hypothesis of Autism (poster from 2016 research conference) Is grandmaternal smoking a force behind the autism surge? (2016 blog piece) Commentary from the Spring 2017 newsletter  With Dr. Temple Grandin, backstage after an autism conference in Northern California. With Dr. Temple Grandin, backstage after an autism conference in Northern California. Last weekend I delivered a talk at an autism conference, among the openers for keynote speaker Temple Grandin (sitting with me at left). After the conference we got to chatting and I mentioned my "time bomb" hypothesis of autism and the idea that grandmaternal smoking, for example, increased the risk of autism in grandoffspring via glitches induced in vulnerable fetal germline. "Well," said the legendary author and new inductee to the Women's Hall of Fame, "my mother's mother smoked like a chimney!"
I have lost count of the number of times I've heard this sort of information about autism grandmothers. When I first started interviewing autism parents about their own prenatal exposures about five years ago, my sole focus was the synthetic steroid hormone "anti-miscarriage" treatments like those to which I had been exposed in utero. Yes I found those hormone stories, but most of the responses went something like this: "No, I doubt my mom took any drugs like that, but she was a heavy smoker," or "My mom smoked a pack a day back when she was pregnant with me," or "Both my mom and mother-in-law were smokers," or "My mother-in-law smoked like crazy," or, of course, "My mother smoked like a chimney." At first I thought nothing of the information, I considered cigarette smoke a mundane and uninteresting fetal exposure compared to the fake hormone protocols my mother was given. But after hearing the smoking story for what seemed like the bazillionth time I did some research to try to connect the dots. And what I found hit me with a wallop: cigarette smoke harbored a long list of toxicants such as benzo[a]pyrene and nicotine, induced mutagenesis and somatic mosaicism, damaged germ cells on many levels, had adverse epigenomic effects on fetal cells, and, from a phenotypic point of view, did direct damage to the fetus, including low birth weight and some evidence of behavioral differences such as ADHD. And there was new evidence from animal models of intergenerational adverse effects of gestational tobacco exposure, including what I would call "mousie ADHD" resulting from nicotine given to the gestating grandma mouse. When paired with the temporality of the autism increase (decades after the parental in utero exposures), the strange socio-demographics of autism, recurrence of ASDs and related pathologies among siblings, and the much-replicated findings of heterogenous de novo mutations, not to mention my somewhat casual surveying of autism families, grandmaternal smoking struck me as a rather formidable hypothesis. When we ponder tobacco toxicity we tend to think of lung cancer, heart disease and stroke, among other somatic pathologies. And sometimes we even think of the millions of fetuses who were heavily exposed in those decades when pregnancy smoking was not only common, but at times prescribed for the purposes of appetite control or anxiety relief. But what about the vulnerable fetal germ cells simultaneously exposed back then during this critical window of germline synthesis? Had anyone looked? To my amazement, the answer was pretty much no. And no autism studies were considering the question either. So I decided to make this question a priority even though it was absent from radar when I began my research quest in 2012. I'm happy to report that some of our projects are now looking at neurodevelopmental outcomes in grandoffspring of women who smoked during pregnancy. Knowing this could be a question of national, or international, importance, I have also contacted about a half dozen PIs of various autism cohorts asking (well, let's be honest, begging) them to inquire about grandmaternal smoking as they do their work evaluating either genomics or proximal fetal exposures, and though I don't yet have any takers, I detect growing openness to the question. One more thing for autism research to consider—the so-called "broader autism phenotype." Are we seeing the BAP in sibs and parents because of natural genetic variation, or at least in part because of acute but forgotten prenatal exposures suffered by us autism parents born during the Mad Men era of maternal medicine? Born in 1965, I was a young subject in a landmark 1977 study finding what we today would call "Aspie traits" in offspring exposed to heavy doses of synthetic progestins. (See the study here: Reinisch and Karow (1977) Prenatal Exposure to Synthetic Progestins and Estrogens: Effects on Human Development). Could the same induced variation hold true of autism parents exposed as fetuses to developmental toxicants such as cigarette smoke (and/or alcohol or drugs or meds)? Well, again, no one has asked. The BAP has so far been presumed to be all natural, a bit of dogma and assumption-making perhaps rooted in ignorance of biological history. In sum, might there be at least one "smoking gun" behind a subset of the autism increase? Perhaps in autism research we should start not with the autistic kids, or even their parents, but with the grandmothers. Jill Escher February 27, 2017 Note: Sometimes I need to pinch my arm to remind myself that it's 2017. You know, it's easy to forget! Because so much of our world is lost in a time warp. Take, for example, the FDA, which seems to be completely — and I mean completely — oblivious to the fact (1) that fetuses have germ cells, and (2) that fetal germ cells are tissues of interest for pharmaceutical toxicology. Basic high school biology stuff. Oh, people of the United States, I weep for all of you, and for all our successive generations. —JE  Escher Fund for Autism
Division of Dockets Management Food and Drug Administration Department of Health and Human Services 5630 Fishers Lane, rm. 1061 Rockville, MD 20852 February 8, 2017 Re: Docket Number FDA-2015-P-0876—Re Citizen Petition to Withdraw Approval for 17α-Hydroxyprogesterone Caproate (“17-OHPC,” Including Brand “Makena”) as a Drug Used in Pregnancy, Pending Fetal Germline Impact Assessment To the Commissioner of the Food and Drug Administration: The FDA received the undersigned’s above-referenced petition on March 19, 2015. Nearly two years later, the agency has taken no action on the petition, which seeks immediate action to withdraw approval for the pregnancy drug 17-OHPC, pending fetal germline impact assessment. The synthetic compound at issue is an endocrine disrupting chemical (“EDC”) widely administered to pregnant women, exposing simultaneously their fetuses and those fetuses’ early germ cells to that chemical and/or its metabolites, as the chemicals easily pass the placenta and enter fetal tissues, including gonadal tissues. EDCs are documented to interfere with healthy germ cell development. By sitting on the petition, the FDA is acting in reckless disregard for the gametic health of countless exposed offspring, and risking the developmental integrity of their offspring, and in so doing, continuing its indefensible and outdated policy of ignoring outright the pharmaceutical risks to vulnerable fetal germline. The petitioner asks that the FDA act expeditiously to grant the petition. • There is little question that germ cells are direct targets of endocrine disruptors from the very early stages of fetal gonad formation. Epigenetic pathways are crucial for germline development and EDCs can interfere with epigenetic mechanisms, including DNA methylation, histone modifications, changes of chromatin structure and microRNA expression, resulting in the transmission of deranged genetic information to the following generation. • In genes subject to fine dosage control such as imprinted genes, for example, geno-affective exposures such as EDCs may easily cause small molecular glitches with catastrophic, life-long neurodevelopmental consequences. • In June 2015, the world’s most esteemed hormone biologists, the Endocrine Society, issued a public statement that EDCs must be assessed for fetal germline impacts. Petitioner is not presenting a “fringe” or novel question, but rather a fundamentally critical issue for drug toxicology and generational public health. • The drug at issue has only a most minimal impact on preterm birth, and patients can receive natural progesterone analogues instead of 17-OHPC where preterm birth is a consideration, while the drug is undergoing safety review. • As an alternative to removing the drug from the market, the FDA can require the vendors of this drug to warn all consumers of the possibility for germline damage to their babies, and to their eventual grandchildren. • In the event that adequate safety assessments reveal no adverse molecular effects on fetal germline, the vendors of 17-OHPC may resume marketing and selling the drug. It is tragedy that the FDA, which is commissioned with the duty to assess damaging effects of pregnancy drugs, seems to possess neither the ability nor interest to contemplate the manifest, unequivocal drug vulnerabilities of fetal germline, even to intentional, high-dose persistent EDC exposures such as 17-OHPC. Given the potential germline consequences of the drug as outlined in the petition, the petitioner implores the agency to act responsibly and grant the petition without further delay. The petitioner wishes again to thank FDA staff for its consideration. Very truly yours, Jill Escher Escher Fund for Autism  Since vaccines have clearly not contributed to the increase in autism, what questions should we should be asking instead? President Donald Trump The White House 1600 Pennsylvania Avenue NW Washington, DC 20500 January 23, 2017 Dear President Trump, As the mother of two children with severe, nonverbal autism, I must give you credit. While most politicians ignore the devastating and costly autism crisis that has swept our country, you have been unafraid to voice your concern. The tremendous surge in serious neurodevelopmental pathology over the past three decades has led to what is now one of our nation’s greatest public health and social services predicaments. And yet we still have few explanations for what lies behind this grim phenomenon. Given the persistent vacuum of information and people’s rather understandable desire to make sense of inexplicable tragedies and hardships playing out in more than a million homes across the states, it’s not terribly surprising that attractive, if superficial, theories have taken root, even in the face of overwhelming evidence against them. One hypothesis, with which you may be flirting, is the idea that vaccines cause autism. Another idea posits that we are all essentially delusional—that autism is perfectly natural, and rising rates are simply an artifact of awareness, broader diagnostics and re-labeling of conditions that once bore other names, such as childhood schizophrenia. The collective data from epidemiological and basic research show emphatically that neither of these views is correct. Vaccines do not cause autism. And the majority of the autism surge cannot be explained by heightened awareness and diagnostic shifts. Nevertheless, in their respective and somewhat diametrically opposed corners, these fictions stubbornly persist. I’m writing this letter because we must move past this dead-end false dichotomy and approach the autism question with both the urgency and biological rationalism it deserves. You and your staff seem to have the urgency, now let’s nail the biological rationalism. Research is increasingly demonstrating that the impairments we call autism are disorders of brain development and function, particularly with respect to circuits that underlie learning, and that the dysfunctions begin to show their hands in early fetal development. While some direct somatic prenatal impacts raise the risk for autism, such as fetal exposure to certain anti-seizure medications, some infections, and prematurity, those factors appear to explain only a fraction of the overall cases. Many researchers are quick to call the unaccounted-for remainder “genetic” mainly because autism is without doubt strongly heritable, meaning in most cases it likely stems from glitches within the egg and/or sperm of the parents, and not from proximal blows to the developing brain (as is the case with Zika microcephaly). Many counter, however, that autism can’t be genetic because genes don’t change that quickly or easily, and moreover we can’t seem to find genetic causes for more than about 10% of autism cases. And that is true—our millenia-old human genes can’t explain the increase in autism, and the DNA-scouring approach has largely come up empty-handed. The problem is this “genes or environment” view of autism is premised on a faulty, incomplete and outdated view of biology, and is therefore misguiding our federal approach to the issue. The past ten years have seen a sea change in how biologists think about heritability, moving away from simple genetic determinism, which still regrettably dominates autism research, and toward a multifactorial systems view embracing the importance of many aspects of genome and gamete biology, in addition to other developmental factors. If we want to understand autism, our research must flow from biological realities, not outmoded hyper-simplistic notions. Here’s a quick biology lesson. DNA is but a single piece of an extraordinarily complex molecular apparatus within gametes that holds sway over later neurodevelopment. Normal brain growth and function depends on a galaxy of itty-bitty on-off switches lying between genes, several “epigenetic” processes, which include chemical tags within and atop DNA helping control gene expression, and cytoplasmic molecules such as tiny RNAs that help define final protein products, among other factors. As the field of genomic toxicology has discovered, these crucial molecular processes can be vulnerable to disruption. With this vulnerability in mind, I seed-fund pilot projects investigating generational impacts of fetal exposure to certain toxicants, including tobacco (via maternal smoking pervasive in the second half of the 20th century), certain pregnancy drugs, and other surprising exposures such as maternal general anesthesia. This work is based in part on my experience discovering I had been exposed in utero to acute doses of powerful synthetic steroid hormone administered to my mother when pregnant with me (and my eggs). I believe my two children’s catastrophically abnormal neurodevelopment likely stems from unforeseen errors laid down by aberrant hormonal signaling in my vulnerable egg precursor cells. I have found many other autism parents with this same exposure story, and many other autism parents with stories of prenatal exposures to other toxicants of interest. This concept has come to be known as the “Time Bomb” hypothesis of autism because it links forgotten gamete exposures in the parents to mystifying pathology in children some decades later. If we accelerate autism research as we should, I believe this hypothesis warrants attention. Another important and related field for research relates to exposures in the pre-conception window. Taken as a whole, studies suggest that subtle impairments in sperm or in the conceptus (perhaps precipitated through certain forms of assisted reproductive technology) may contribute to autism risk. In one study, for example, abnormal epigenomic marks were found in the sperm of fathers of children with autism. It’s critical to better understand what exposures or manipulations may perturb the late-stage gametes and elevate the risk for dysregulated neurodevelopment. A third area for improved research involves direct effects on central nervous system development. Some studies suggest that early exposure to certain chemicals, including endocrine-disrupting compounds, may have adverse and persistent effects on healthy brain development. Though some work is being done in this field already, there are many chemicals that warrant more thorough review for developmental safety. While vaccines—like all pharmaceutical drugs—carry some risks, there is no reason to attribute the stunning surge in autism to them. Allow me to offer a few of the reasons:
Mr. President, you are an expert businessman. You do not waste time and you do not waste money. But further inquiry into the autism/vaccine connection is a waste of time and money at exactly the moment we should be urgently hunting down more promising theories, some of which are described above. Americans deserve answers, and this is no time for either complacency (autism is all “natural diversity”) or unscientific dead-ends (vaccine blaming). I believe considerable progress on autism is firmly within our grasp. Thank you for your sincere concern about the autism crisis, which deserves a place as one of America’s top policy priorities, and your consideration of these ideas. Very truly yours, Jill Escher Jill Escher is founder of the Escher Fund for Autism, the mother of two children with nonverbal forms of autism, an active advocate for autism causes in the San Francisco Bay Area, and a housing provider to adults with autism and developmental disabilities. Learn more about her hypothesis at the Escher Fund’s science education website, www.GermlineExposures.org.  We are pleased to announce the addition of five fascinating new expert interviews at GermlineExposures.org.
Toshi Shioda, MD, PhD, Harvard University: A Revolution in Germline Toxicology: Primordial Germ Cell-Like Cells (PGC-LCs) One of the barriers to understanding germline exposure risk is the lack of effective and reliable models for mechanistic studies. Dr. Shioda’s lab has taken advantage of cutting edge tools from stem cell biology and deep sequencing technology to develop germ cell models on which new toxicological tests could be run. "If epigenetic errors occur in the germline genome during the reprogramming process during pregnancy of a woman, her sons or daughters appear normal, but their germline cells carry a potential bomb." Piroska Szabó, PhD, Van Andel Institute: Chemicals Can Exert Direct Epigenetic Effects on Exposed Fetal Germ Cells Dr. Szabó's recent paper in Genome Biology reported that endocrine disruptors affected the global transcription and DNA methylation state of exposed fetal mouse germ cells, but these aberrations were not passed on to the germ cells of the subsequent generation. "I am concerned about harming the exposed germ cells by the chemicals we have tested. I also feel very concerned about potentially harming the germ cells by the many thousands of additional man-made chemicals that humans or wildlife can’t avoid being exposed to." Miklos Toth, PhD, Cornell Medical Center: Non-DNA Mediated Transmission of Behavior Across Generations Maternal factors during sensitive periods of development produce persistent effects in the offspring, including effects on brain development and behavior. Dr. Toth's latest paper looks beyond fetal effects to successive generations, looking at altered methylation and gene expression in genes whose functions can be linked to behavioral traits, and ultimately, changes in neuronal function and behavior in generations of mice. "We believe that iterative somatic transmission (through bioactive compounds via the placenta and breast milk) of behavioral traits is a prominent intergenerational and multigenerational mechanism." June Reinisch, PhD, Prenatal Development Project, and former director, The Kinsey Institute: The "Hidden History" of Pregnancy Drugs in the Postwar Era Prenatal Exposures Oral History Project Dr. Reinisch is renowned for her pioneering investigations of adverse impacts of various post-war pregnancy drugs, including once-common synthetic steroid hormones and barbiturates, on fetal development. In the 1970s Jill Escher, who had been exposed in utero to large quantities of synthetic steroid hormones, was one of her study subjects. "There was a gigantic growth in all kinds of pharmaceuticals after World War II. Since there was this idea of the fetal placental barrier, there was the notion you could treat the mother without interfering with the baby. That went on for much longer than it should have." Mark Klebanoff, MD, MPH, University of Ohio: Postwar Obstetric Practices Prenatal Exposures Oral History Project Dr. Klebanoff has examined a variety of outcomes in cohorts of pregnancies from the post-war decades, during which some myth-driven obstetric practices were the norm. "It gave me a good healthy dose of humility because the between-the-lines message was let's wait another 40 years to find out how many things we do today do more harm than good." See all 35 expert interviews: http://www.germlineexposures.org/expert-qa.html  [This post was written by my friend, a DES victim named Marcia Love. It was written for her blog IWantAnApology.com. While I was not a DES daughter, I was exposed in utero to heavy amounts of other synthetic steroid hormones, and I identify with her plight and outrage at the lack of accountability in the medical and regulatory systems regarding this violently toxic pregnancy drug that was given recklessly to millions of gestating women. I'm proud to say I am now sponsoring a study on grandchild (germline) effects of DES on development of those offspring of exposed gametes. -Jill Escher]
They Made Us Freaks And Gave Us Cancer! The True Story Of The Largest Disaster In Pharmaceutical History by Marcia Love This page is about the drug Diethylstilbestrol. When you think about the largest or greatest drug disaster most people think of Thalidomide which was certainly a devastating catastrophe. The heartbreaking pictures of those born without limbs or those with deformed limbs can never be forgotten by those who have seen them. In terms of the number of people affected Diethylstilbestrol was far worse. Th CDC estimates that over ten million people were exposed. There are a few very profound differences between the two drugs. First, the physical defects from Thalidomide were very apparent and immediate. A baby born with no limbs draws attention. One born with a 40x increased risk of cancer 20 years down the road not so much. Next we have the fact that Thalidomide was not distributed worldwide so the number of people exposed was far less. The number of people exposed to Thalidomide is in the tens of thousands. Then there is the fact that Thalidomide is still being sold and in fact many studies have for instance shown it to be effective in the treatment of multiple myeloma, especially when used with other drugs. And of course Thalidomide was never sold in the U.S. For these reasons every scientist on the planet and most lay men as well know about Thalidomide but very few know about Diethylstilbestrol. Know that the pharmaceutical industry here in America operates the same way the tobacco companies did foryears. Deny responsibility, Deny your product kills and pay shills to promote your product in the face of increasing evidence which says that it indeed kills. In 2012 Gruenenthal Group (The company which sold Thalidomide) to mothers and their children for the tragedy. No such apology has ever been offered for Diethylstilbestrol. In 2010 then senators John Kerry and Scott Brown sent a letter asking for an apology from the FDA. The response they got was something like “we are making sure that doesn’t happen again.” They never got their apology… Diethylstilbestrol was discovered in England at Oxford University in 1938 by Sir Charles Dodds. It was found to have have estrogen like effects and was marketed as a cheap synthetic replacement for estrogen. The supposed premise that it was sold on was that since it behaved like estrogen it could be used as a cheap synthetic estrogen. As it turns out Diethylstilbestrol is not a synthetic estrogen but an endocrine disruptor. Dodds warned of intersex and cancerous outcomes and the FDA initially denied approval of the drug. This did not stop the pharmaceutical companies from aggressively campaigning to get the drug approved. The reason? It was a free drug that anyone could manufacture and sell. English lay at the time forbade money to be made on anything discovered with public funds. The drug companies pooled their resources and formed what was known as the Small Committee, hiring former Eli Lilly CEO Don Carlos Hines to represent them. Soon there was an article in the Readers Digest stating that a new wonder drug for treating post-menopausal symptoms was available and was just awaiting the approval of the FDA. Such articles asked readers to send letters to their congressmen and even the president. The aggressive campaign worked and by 1940 the FDA had approved it for use in treating gonorrheal vaginitis, menopausal symptoms, atrophic vaginitis and lactation suppression. The gonorrheal vaginitis indication was removed when penicillin was discovered. At the behest of their pharmaceutical representatives doctors also began prescribing Diethylstilbestrol off label for the purpose of preventing miscarriages. As with the other four approved indications no proof was ever provided as to the efficacy of the drug for this purpose. Scientific method was not widely used at this point in history because the FDA did not yet require it even though it was standard practice as early as the late 1930’s. So the MD was easily convinced by the affable drug rep to prescribe it for his at risk patients without any proof that it actually worked. Off label prescription of Diethylstilbestrol for preventing miscarriage ended in 1947 when the FDA approved it for that use after years of pressure from the pharmaceutical companies. They convinced doctors and patients alike to write letters attesting to the safety and effectiveness of the drug. Then came George and Olive Smith two “researchers” from Harvard who began advocating for an increasing regimen ending at 125 mg/day during the final month of pregnancy. At the time Eli Lilly was selling it in doses of .1,.5 and 5 mg. The most outrageous thing is that there was absolutely no proof of efficacy of these large doses because the scientific method had yet to be employed on the drug! So then Bristol-Myers Squibb sought and and was granted permission to market and sell Diethylstilbestrol in doses of 25 mg and 100 mg. As a fetus develops it’s hormonal needs vary by the micro-second and it is impossible for anyone to know what those exact needs are. DES is a powerful endocrine disruptor and introducing it into the womb at random times leads to arbitrary results. Some women ended up with a T-Shaped uterus, some did not. Some sons became transgender, some did not. There are commonalities in mothers, sons and daughters however. Finally in 1951 a double blind study was conducted at the University of Chicago and Diethylstilbestrol was proven to have to no effect on the prevention of miscarriages. This fact did not slow down sales however. Coming from someone who was born in 1956 and exposed to the Smith & Smith regimen you can imagine how heartbreaking that fact is. The person I was meant to be was never born and my body was permanently altered before I was born for no reason other than profit. Study after study proved that Diethylstilbestrol did absolutely nothing to prevent miscarriages but yet the FDA still allowed it to be sold. By the late 1960’s six of the seven leading textbooks on Obstetrics and Gynecology said Diethylstilbestrol had no value in preventing miscarriages but it wasn’t until 1971 when the New England Journal of Medicine published a study which found an astounding link between vaginal clear cell adenocarcinoma and in utero exposure to Diethylstilbestrol in women and young girls. Prior to this that type of cancer was only found in older women and even then only rarely. Even after it was proven to cause cancer the FDA only removed prevention of miscarriage as in indication for Diethylstilbestrol use and added pregnancy as a contraindication for Diethylstilbestrol use. The FDA never banned DES outright and other indications were allowed to stand but as evidence mounted the FDA started taking more action against it and in 1975 ordered 25mg and 100mg tablets withdrawn from the market. In 1978 the FDA removed postpartum lactation suppression from their approved indications and by the 1990’s the only approved indications were for advanced breast cancer in postmenopausal women and for the treatment of advanced prostate cancer. Finally in 1997 the last U.S. manufacturer Eli Lilly stopped making and marketing Diethylstilbestrol. For 60 years Diethylstilbestrol was prescribed for made up reasons without any scientific data whatsoever to warrant it’s prescription. If it wasn’t so heartbreaking it might be laughable some of the reasons it was prescribed. Starting in the 1950’s and continuing through the 1970’s Diethylstilbestrol was prescribed for “excessive height” in prepubescent girls, particularly in Australia. The CDC estimates that 10 million people were exposed between 1938 and 1971 the vast majority for the prevention of miscarriage ruse. That means 5 million DES mothers gave birth to 2.5 million DES sons and 2.5 million DES daughters. From the CDC’s website: “More than 30 years of research have confirmed that health risks are associated with DES exposure. However, not all exposed persons will experience the following DES-related health problems.
In addition to causing cancer Diethylstilbestrol is a known teratogen, that is it is capable of causing physical malformations to those exposed in utero. DES exposed daughters have a higher risk of reproductive tract abnormalities including epithelial cell changes known as vaginal adenosis, an increased cervical transformation zone and uterine abnormalities such as a T-shaped uterus. In 2005 Dr Scott Kerlin published the results of an on-line survey of DES sons. Out of 500 surveyed 150 men reported gender identity issues. Looking at the math: 150/500 x 2.5 million = 750,000 male to female transexuals were artificially created by the pharmaceutical industry. Estimates of the natural occurrence of transexualism was somewhere between 1 in 5,000 to 1 in 70,000 depending on who you believe. Sill noting compared to the 3 in 10 you get when administering DES into the womb. I am a DES son and there is virtually no help out there for me. I have a microphallus, ovaries, intermittent gender dysphoria and a whole lot more. I am demanding an apology from the FDA, Eli Lilly and Bristol-Meyers Squibb and Harvard University. Because of their blind pursuit of profit, ignoring all scientific proof of the harm their products do to developing fetuses and refusing to take responsibility I and millions of others have suffered greatly and have led miserable lives. Their greed has caused me life-long pain and humiliation and ensured I would never find the happiness our constitution allows us to pursue. It is time to fight back… IwantAnApology.com Letter to FDA's Center for Tobacco Products Regarding Study of Tobacco Smoking Impacts on Germ Cells10/17/2016 Escher Fund for Autism
Mitch Zeller Director, Center for Tobacco Products FDA Document Control Center 10903 New Hampshire Avenue Building 71, Room G335 Silver Spring, MD 20993-0002 October 17, 2016 Re: Center for Tobacco Products Efforts to Address Tobacco-Induced Human Germ Cell Damage Dear Mr. Zeller, I am a science philanthropist primarily concerned about adverse mutagenic and epimutagenic effects of various drugs, pharmaceuticals and tobacco on the human germline. Based on my admittedly partial review of CTP activities, it appears that the CTP limits its concern for adverse outcomes of tobacco exposure to the somatic level. While questions of somatic pathology such as lung cancer, addiction, and heart disease are clearly of utmost importance in safeguarding public health, they also miss an entire dimension of risk. Owing to its various toxic components, tobacco smoke has been shown to cause adverse impacts on germ cells. It has been shown to induce mutation in germ cells in human studies and animal models, as well as somatic mosaicism in animal models. At least one study points to transgenerational neurodevelopmental effects of nicotine, most likely via epigenetic mechanisms. The mutagenic and epimutagenic properties of tobacco smoke are now well known. It is therefore surprising that the FDA’s CTP does not seem to include human germ cells, both male (sperm and precursors) and female (eggs and precursors), as a potential endpoint for tobacco toxicity. If in fact the high pregnancy smoking rates of the past decades increased risk for germline mutation and/or epimutation, developmental abnormalities in resulting offspring may be significant, and may include neurodevelopmental disorders such as autism and ADHD. I am writing to inquire about what you think could be done at the CTP to ensure the FDA expeditiously addresses tobacco-induced risks to germ cells, particularly the highly vulnerable early germ cells that lie within fetuses. Health Canada, for example, has an admirable program on the gametic genetic toxicology of tobacco. What can we do to ensure a vigorous response here in the United States at the FDA as well? Thank you for your consideration. Very truly yours, Jill Escher Escher Fund for Autism GermlineExposures.org |
AuthorJill Escher, Escher Fund for Autism, is a California-based science philanthropist and mother of two children with severe autism, focused on the question of how environmentally induced germline disruptions may be contributing to today's epidemics of neurodevelopmental impairment. You can read about her discovery of her intensive prenatal exposure to synthetic hormone drugs here. Jill is also president of Autism Society San Francisco Bay Area. Archives
July 2021
Categories |

- Home
-
Expert Q&A
- Eva Jablonka Q&A
- Amander Clark Q&A
- Mirella Meyer-Ficca Q&A
- Janine LaSalle Q&A
- Dana Dolinoy Q&A
- Ben Laufer Q&A
- Tracy Bale Q&A
- Susan Murphy Q&A
- Alycia Halladay Q&A
- Wendy Chung Q&A
- Pradeep Bhide Q&A
- Pat Hunt Q&A
- Yauk and Marchetti Q&A
- Emilie Rissman Q&A
- Carol Kwiatkowski Q&A
- Linda Birnbaum Q&A
- Virender Rehan Q&A
- Carlos Guerrero-Bosagna Q&A
- Randy Jirtle Q&A
- Jerry Heindel Q&A
- Cheryl Walker Q&A
- Eileen McLaughlin Q&A
- Carmen Marsit Q&A
- Marisa Bartolomei Q&A
- Christopher Gregg Q&A
- Andrea Baccarelli Q&A
- David Moore Q&A
- Patrick Allard Q&A
- Catherine Dulac Q&A
- Lucas Argueso Q&A
- Toshi Shioda Q&A
- Miklos Toth Q&A
- Piroska Szabo Q&A
- Reinisch Q&A
- Klebanoff Q&A
- Denis Noble Q&A
- Germline in the News
- Science
- Presentations
- About Us
- Blog

 RSS Feed
RSS Feed
Proudly powered by Weebly